Publié le 25 juil 2010Lecture 5 min
Xanthogranulomes juvéniles
D.SCHROEDER, E.MAHE - CHU Ambroise Paré, Boulogne-Billancourt
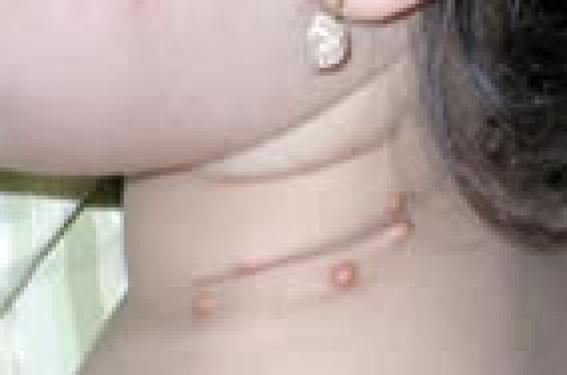
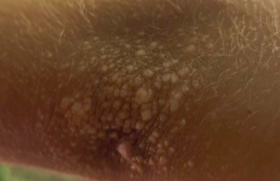
Observations ● Observation 1. Benjamin, 18 mois, présentait depuis l’âge de 6 mois une lésion orangée rétro-auriculaire droite. La lésion avait initialement augmenté de volume, puis était devenue stable avec un aspect orangé légèrement télangiectasique en périphérie, en regard d’un nodule sous-cutané de 13 mm de diamètre. Depuis un an, l’augmentation de la taille de la lésion se faisait proportionnellement à la croissance de Benjamin. Le reste de l’examen était sans particularités. ● Observation 2. Sara, 3 ans, se présentait en consultation pour l’apparition un mois avant de multiples papules orangées disséminées. L’examen retrouvait plus de 50 papules orangées localisées sur tout le corps (figures 1 et 2) ainsi que sur le cuir chevelu. Figure 1. Multiples papules orangées cervicales chez une fillette de 3 ans. Elle n’avait pas d’antécédents notables et était en excellent état général. L’examen, outre ces papules était strictement normal. Il n’était notamment pas retrouvé d’organomégalie. L’examen ophtalmologique et l’hémogramme étaient normaux. Diagnostic retenu Devant ces papules jaunâtres apparaissant d’emblée chez des enfants en excellent état général, le diagnostic de xanthogranulomes juvéniles (XGJ) a été retenu dans les deux cas, XGJ unique chez Benjamin et multiples chez Sara. Le XGJ de Benjamin était légèrement atypique par le caractère très nodulaire initial. Cependant, la surveillance clinique était rassurante, la lésion restant stable. Une simple surveillance clinique annuelle a été instaurée. Pour Sara, le diagnostic de XGJ multiples a imposé une surveillance plus rapprochée. L’examen ophtalmique a éliminé une localisation oculaire, seule localisation pouvant poser des problèmes fonctionnels. L’hémogramme a éliminé une leucémie pouvant exceptionnellement être associée à ces XGJ multiples. Après 2 ans de surveillance, les lésions ne se sont pas multipliées, et les premiers XGJ apparus ont eu tendance à disparaître en laissant des cicatrices atrophiques. Commentaires La xanthogranulomatose juvénile est la forme la plus commune d’histiocystose non langheransienne et survient le plus souvent chez le nouveau-né ou le nourrisson. Elle n’est pas en rapport avec une perturbation du métabolisme lipidique. Les lésions correspondent à des papules ou des nodules de 5 à 10, voire 20 mm de diamètre, de couleur jaune, orange ou brune, de consistance molle, associées parfois à des télangiectasies en superficie. La lésion est souvent unique (dans 60 % des cas), mais peut être multiple. Figure 2. Papules orangées du tronc chez la même fillette. À la phase initiale, l’aspect orangé peut ne pas être retrouvé, la lésion est alors plus inflammatoire et peut faire suspecter d’autres types de tumeurs bénignes ou malignes. C’est l’évolution, ou l’histologie dans les cas les plus suspects, qui permettra de redresser le diagnostic. La plupart du temps asymptomatique, elle siège préférentiellement au niveau de la tête, du cou, du cuir chevelu, du tronc et de la racine des membres. Le pronostic est bénin avec régression spontanée des lésions en 3 à 6 ans chez l’enfant. Elles peuvent laisser une cicatrice anétodermique. Le pronostic est bénin avec régression spontanée des lésions en 3 à 6 ans chez l’enfant. Diagnostics différentiels Devant un aspect typique, peu de diagnostics peuvent être discutés, principalement la xanthomisation d’autres lésions bénignes. En cas de doute diagnostique, la biopsie cutanée retrouvera un infiltrat dermique dense, bien limité, fait d’histiocytes spumeux et de cellules géantes, dont le nombre peut varier en fonction de l’âge des lésions (moins nombreuses si lésions très précoces ou tardives). L’analyse immunohistochimique présente un intérêt pour éliminer les diagnostics différentiels. Dans les XGJ, les histiocytes sont négatifs pour la protéine S100 et le CD1a, et positifs pour la vimentine et le CD68. On éliminera ainsi les autres proliférations histiocytaires : – histiocytose de Hashimoto- Pritzker, de régression plus rapide que la XGJ, ou l’histiocytose céphalique bénigne ; – des proliférations mélanocytaires pouvant avoir un caractère légèrement orangé telles que le nævus de Spitz ; – il faut également éliminer les exceptionnelles tumeurs cutanées malignes pouvant avoir cet aspect orangé caractéristique du XGJ : le rhabdomyosarcome, le fibrosarcome et le fibrohistiocytome malin. Prise en charge Il n’y a pas de traitement spécifique de la lésion. Elle va régresser spontanément. Une exérèse chirurgicale peut être proposée en cas de localisation « gênante » socialement, au prix d’une cicatrice obligatoire. L’examen clinique doit être attentif, à la recherche d’une atteinte extracutanée : – atteinte ophtalmique : principale localisation extracutanée du XGJ, mais qui reste rare : 0,3 à 0,5 % des XGJ. Cette localisation peut entraîner un hyphéma, un glaucome, une uvéite, une atteinte palpébrale… La recherche d’une atteinte ophtalmique est justifiée en cas de forme diffuse ou de signes d’orientation. Chez l’enfant de moins de 2 ans porteur de XGJ multiples, un examen ophtalmologique est indiqué de façon systématique tous les 6 mois jusqu’à l’âge de 2 ans, en raison du risque accru d’atteinte oculaire ; les signes ophtalmo- logiques devront être également expliqués aux parents ; – les localisations pulmonaires (dyspnée, opacités radiologiques) ou hépatiques (hépatomégalie) sont beaucoup plus rares et à rechercher en cas de point d’appel clinique ; – d’autres localisations ont été exceptionnellement rapportées : pancréas, cervelet, rate, testicules et péricarde. En présence de XGJ multiples, l’examen clinique et l’interrogatoire devront s’attarder à rechercher particulièrement des éléments en faveur d’une neurofibromatose de type 1, dont l’association ne serait pas exceptionnelle. L’apparition de XGJ chez un patient porteur de neurofibromatose de type 1 doit amener à réaliser une histologie et à effectuer une surveillance clinique et biologique rapprochée compte tenu du risque de survenue d’une leucémie myélomonocytaire chronique juvénile.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :