Publié le 18 déc 2007Lecture 9 min
Un syndrome abdominal douloureux trompeur
D. ARMENGAUD - Hôpital intercommunal, Poissy/Saint-Germain-en-Laye
Thibaut, petit garçon de 14 mois et demi, présente depuis la veille au soir des difficultés d’alimentation, avec douleurs d’allure abdominales mises sur le compte d’une gastro-entérite du fait de l’apparition concomitante de deux selles liquides. Malgré l’adaptation du régime alimentaire, les parents se décident à venir aux urgences car l’enfant refuse toute alimentation et présente des pleurs quasi incessants qu’ils n’arrivent pas à calmer.
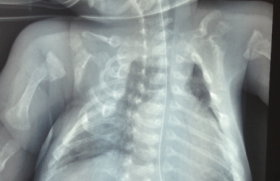
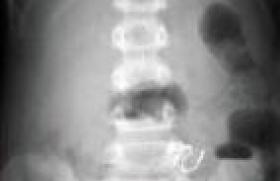
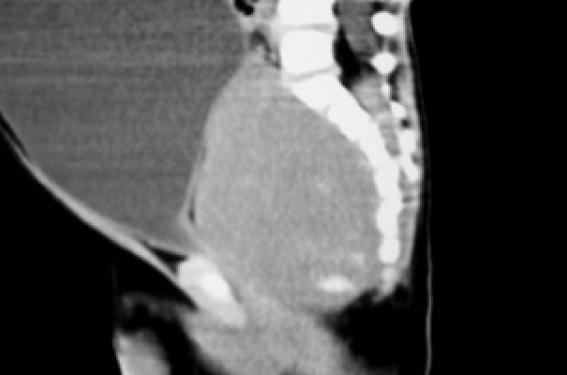
Histoire clinique Il s’agit d’un premier enfant d’un couple sans antécédents particuliers. La grossesse s’est déroulée normalement et l’accouchement a eu lieu à 38 SA par césarienne en raison d’une stagnation de la dilatation. Le poids de naissance était de 3 220 g la taille de 49 cm et le PC de 32 cm. À l’examen, l’enfant paraît algique et geignard, ne se calmant pas effectivement dans les bras de ses parents. La température est notée à 38,5 °C, la fréquence cardiaque est de 144/min, la pression artérielle est de 83/59 mmHg et le poids de 10 kg. L’abdomen est distendu paraissant plus souple à gauche qu’à droite, où la palpation semble globalement douloureuse et trouve une masse rénitente plutôt hypogastrique. Le foie est palpé, débordant de 1 cm le rebord costal ; il n’y a pas de splénomégalie et les fosses lombaires sont libres. Le transit semble conservé (une selle liquide est émise pendant l’examen), et si l’enfant ne s’alimente pas beaucoup, la prise d’une solution de réhydratation orale au biberon est effective. L’auscultation pulmonaire est normale en dehors de quelques râles bronchiques. L’examen ORL est normal, tout comme l’examen neurologique (marche acquise quelques semaines auparavant). Il n’y a pas d’éruption . Un bilan biologique est prélevé, avant d’adresser l’enfant en radiologie pour un ASP (figure 1). Examens biologiques NFS – Hb : 10,7 g/100 ml – GR : 4 050 000/mm3 ; (VGM 74 µ3) – GB : 15 300/mm3 (68 % de PNN) – Plaquettes : 319 000/mm3 Ionogramme sanguin (mmol/l) : Na : 129 ; K : 4,7 ; bicarbonates : 15 ; urée : 22,8 Protides : 71 g/l Calcémie : 2,25 mmol/l Créatinine : 188 µmol/l ASAT : 50 UI/I (N : 15-40) ; ALAT : 29 UI/I (N : 15-70) CRP : 58 mg/l Figure 1. Abdomen sans préparation. Masse abdominale médiane, refoulant les anses digestives en périphérie et s’étendant jusqu’au petit bassin. L’enfant se présente avec un tableau abdominal pouvant faire évoquer au premier abord une occlusion par invagination intestinale aiguë ou un volvulus du fait des troubles digestifs et de la distension abdominale. Cependant, la biologie est d’emblée inquiétante devant la mise en évidence d’une insuffisance rénale avec une augmentation de l’urée à 15 mmol/l, que n’explique pas la clinique car il n’y a pas de signes de déshydratation ; l’augmentation de la créatinine à 180 µmol/l est en faveur d’une origine organique (rénale) et non fonctionnelle (prérénale). Pour autant, le reste de la biologie n’apporte pas d’éléments d’orientation supplémentaire ; un syndrome hémolytique et urémique est éliminé sur l’absence d’anémie et de thrombopénie. L’ASP confirme la présence d’une masse abdominale médiane refoulant les anses digestives sans signes directs ou indirects d’occlusion. Quelle hypothèse doit-être émise et quel examen demander ? Il est évident que l’imagerie doit être poursuivie par une échographie et/ou un scanner qui pourront être réalisés successivement. Échographie abdominale (figures 2). Masse liquidienne correspondant à un volumineux globe vésical avec masse médiane sous jacente à l’origine de la compression (prostate ?). Figure 2. Échographie abdominale. (a) La masse abdominale palpée est liquidienne et correspond à un globe vésical. (b) Distension des cavités pyélocalicielles secondaires au globe vésical obstructif, probablement à l’origine de l’insuffisance rénale aiguë. Tomodensitométrie (figures 3). Le col vésical est refoulé très en avant derrière la symphyse pubienne par une masse qui n’est pas prostatique, plus postérieure et semblant contenir des calcifications. Il existe un retentissement important sur les voies urinaires hautes. Il semble exister une extension foraminale et endocanalaire au niveau du sacrum (composante en sablier). Figure 3. (a) TDM sans injection. Globe vésical et distension des cavités pyélocalicielles. (b) TDM avec injection. Coupe sagittale montrant le globe vésical et la présence en arrière d’une masse rétroprostatique comprimant l’urètre contre la symphyse pubienne. Noter la présence de calcifications dans le petit bassin. Discussion diagnostique Il s’agit donc de la révélation par une compression vésicale d’une tumeur du petit bassin pouvant faire évoquer deux ou trois diagnostics : – un tératome sacro-coccygien, mais si la tumeur semble contenir quelques calcifications, elle est homogène ; – un rhabdomyosarcome, mais son origine dans le petit bassin pourrait en faire un point de départ prostatique qui semble ici exclu par l’imagerie ; – mais surtout et en premier lieu à cet âge, un neuroblastome pelvien, ce d’autant qu’il existe des calcifications et un doute sur une composante canalaire. Dans un premier temps la pose d’une sonde urinaire permettra l’évacuation d’un volumineux globe vésical, ramenant environ 500 ml !!! d’urines claires non hématiques. Ce geste permet, associé à la mise en place d’une perfusion veineuse périphérique de base, la récu-pération rapide dans les jours suivants d’une fonction rénale normale (créatinine : 63 µmol/l ; urée : 2,2 mmol/l), sans troubles de concentration des urines. L’enfant est alors transféré en milieu spécialisé pour la poursuite de la prise en charge. Le diagnostic de neuroblastome sera confirmé par la réalisation, en milieu spécialisé, d’une exploration complète avec : – une biopsie qui mettra en évidence un neuroblastome peu différencié avec un index mitotique (MKI) faible de l’ordre de 2 % ; – une biopsie médullaire ; – une scintigraphie à la méthyl iodo benzyl guanidine (MIBG) ; – un profil génomique (amplification Nmyc négatif). Après 4 cures de chimiothérapie associant vincristine, étoposide et carboplatine, une réduction tumorale de près de 90 % est obtenue permettant la réalisation d’une exérèse chirurgicale qui sera incomplète du fait du caractère adhérent de la tumeur résiduelle au rectum et à la région présacrée (avec persistance de 5 % environ de cellules tumorales dans la pièce d’exérèse). Une chimiothérapie complémentaire s’avère nécessaire. Commentaires Le neuroblastome est une tumeur embryonnaire de début périnatal, développée aux dépens du système nerveux sympathique. C’est la plus fréquente des tumeurs solides extracrâniennes de l’enfant : elle représente environ 10 % des cancers de l’enfant, survenant principalement chez le nourrisson. Le mode de révélation est le plus souvent la découverte fortuite d’une masse abdominale (lors d’un examen systématique) ou thoracique (lors d’une radiographie pulmonaire). Mais en fonction de ses multiples localisations possibles, elle peut être révélée par une complic-ation : compression médiastinale d’un neuroblastome thoracique ; syndrome de Claude Bernard Horner d’un neuroblastome cervical ; para-parésie d’un dévelop-pement intracanalairel ; hépatomégalie et/ou tumeurs cutanées d’une forme IV S ; ou encore manifestations osseuses (douleurs, fracture pathologique) des formes métastatiques (60 % au diagnostic). La survenue précoce, la gravité du pronostic, le risque thérapeutique important chez le nourrisson, l’existence de marqueurs biologiques (cf. encadré) ont fait discuter la mise en place de programme de dépistage basé sur le dosage urinaire des catécholamines. Ces études menées au Japon, au Canada et dans certains pays d’Europe ont malheureusement été abandonnées dans la mesure où elles n’ont pas fait la preuve de l’amélioration globale du pronostic : – possibilité de faux positifs et de faux négatifs ; – conditions particulières de recueil des urines ; – dépistage des formes les moins sévères ; – évolutions parfois spontanément résolutives sous forme de maturation des neuroblastes en neurone ou en cellule ganglionnaire mature ; – les neublastomes non sécrétants immatures sont de plus mauvais pronostic. La vigilance clinique, au cours des premières années de vie, et lors des multiples examens cliniques des nourrissons, reste toujours de mise. Marqueurs diagnostiques et pronostiques du neuroblastome Marqueurs diagnostiques Dans 95 % des cas, le neuroblastome est dit sécréteur et le recueil des urines permet d’évaluer le taux des catécholamines urinaires (acide homovanillique [HVA], acide vanillylmandélique [VMA] et dopamine), dont l’excrétion augmentée est fortement évocatrice du diagnostic. Mais 5 % des neuroblastomes, plus indifférenciés, sont « non sécréteurs ». Marqueurs pronostiques Ils sont principalement utilisés dans le but de mieux adapter les protocoles thérapeutiques : Biologiques (et de plus mauvais pronostic) : – LDH sérique ¼ – ferritinémie ¼ Scintigraphiques. L’accumulation de MIBG dans les cellules catécholaminergiques permet de repérer une tumeur primitive non accessible en imagerie (stade IV S), ou de mettre en évidence des métastases (osseuses). Histologiques (et de plus mauvais pronostic) : – neuroblaste indifférencié, absence de développement de stroma (cellule de Schwann), aspect nodulaire ; – MKI (mitosis karyorrhexis index) > 200/5 000 cellules étudiées ; – immuno-histochimie : NSE ¼ (neuron specific enolase). Génétiques – L’expression exagérée (amplification) du gène Nmyc est certainement le facteur pronostique le plus important . Il s’agit d’un oncogène présent sur le chromosome 12 dont la surexpression est en rapport avec une capacité de prolifération plus importante de la tumeur. – Le gène Hras serait, lui, plutôt associé à des tumeur de stade inférieur et de moindre capacité proliférative. – La délétion du bras court du chromosome 1 (1p36) est souvent associée à l’amplification de Nmyc, mais d’autres délétions ont également été décrites (11q, 14q, 17q). – L’expression de gènes produisant des tyrosine kinase (Trk) est un autre élément d’appréciation, mais si l’expression de la TrkA est inversement corrélée à l’expression du gène Nmyc, et donc de bon pronostic, c’est l’inverse pour TrkB. – L’hyperdiploïdie, en revanche, est plutôt l’indice d’une meilleure chimiosensibilité des cellules tumorales.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :