Publié le 14 mar 2018Lecture 5 min
Ostéogenèse imparfaite : à propos d’un cas
Mohamed KMARI et coll.*, Service de pédiatrie, Hôpital militaire d’instruction Mohammed V, Rabat (Maroc)
Il s’agit d’un nouveau-né, de sexe masculin admis au service dans un tableau de détresse respiratoire apparu à J7 de vie. Il est issu d’une grossesse mal suivie menée à terme, de parents non consanguins, sans pathologie héréditaire connue dans la famille. La mère est âgée de 38 ans, G2P2, sans antécédent particulier. Le père est âgé de 51 ans. Le nouveau-né a deux sœurs bien portantes.
Clinique
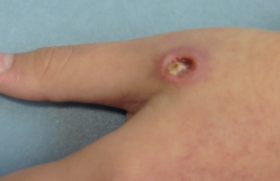
L’examen clinique objective une hypotrophie, des membres courts, une déformation thoracique avec un thorax étroit et un chapelet costal, pied bot droit et fontanelle antérieure baillante, avec une ecchymose au niveau de la face antérieure du thorax et des sclérotiques bleues (figures 1 et 2).
Figure 1. Aspect de sclérotiques bleutées.
Figure 2. Aspect des membres courts.
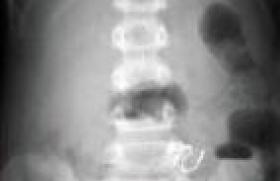
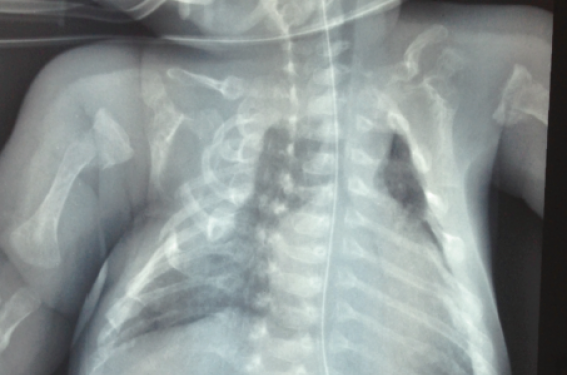
L’imagerie des membres, du thorax et du crâne a montré une transparence excessive du squelette osseux et des corps vertébraux, des incurvations diaphysaires au niveau du fémur avec un aspect grêle, des traits de fractures avec cals vicieux, élargissement transversal du crâne, défaut d’ossification de la voûte crânienne et amincissement cortical (figures 3 et 4).
Figure 3. Radiographie standard montrant des traits de fractures, une déformation thoracique, un humérus incurvé et une transparence excessive du squelette.
Figure 4. Radiographie du membre inférieur montrant un fémur incurvé, avec aspect de cal vicieux et trait de fracture.
Au scanner cérébral, il existe un important amincissement de la voûte occipitale et temporale avec un parenchyme cérébral normal (figure 5). L’échographie abdominale et cardiaque sont sans anomalies. Le bilan phosphocalcique et les phosphatases alcalines sont normaux. Devant l’aspect du malade et les données de l’imagerie, le diagnostic retenu était celui d’une ostéogenèse imparfaite dans sa forme sévère. La maladie est expliquée aux parents avec les mesures de prise en charge à domicile pour éviter les fractures et un contrôle clinique tous les mois. Un traitement par biphosphonates est prévu à 9 mois de vie et un suivi orthopédique en parallèle.
Figure 5. Scanner cérébral avec un amincissement de la voûte crânienne, accentué au niveau de la région occipitale et temporale.
Discussion
L’ostéogenèse imparfaite (OI) est une maladie génétique caractérisée par une fragilité osseuse et une faible masse osseuse : c’est une ostéoporose congénitale(1). Cette pathologie est rare. Sa prévalence est estimée à 1 pour 10 000 à 20 000 personnes, sans facteur racial, ni ethnique particulier(2). Il s’agit d’une affection héréditaire due à une anomalie de production du collagène de type 1, qui peut être quantitative ou qualitative. La majorité des sujets ont une mutation de l’un des deux gènes qui codent pour les chaînes alpha du collagène de type I (COL 1A1 et COL 1A2). Les molécules de collagène sont formées par 3 chaînes polypeptidiques : 2 chaînes identiques α1 codées par le gène COL 1A1 situé sur le chromosome 17 et une chaîne α2 codée par le gène COL 1A2 situé sur le chromosome 7(1). Les trois chaînes constituent une triple hélice, dont le centre ne peut être occupé que par la glycine. Toute substitution par un AA entraîne une déformation importante avec ralentissement des fibres de collagène. Sur le plan génétique, elle est de transmission autosomique dominante. Cependant, des cas sporadiques ont été rapportés d’enfants malades de parents non affectés et des cas de transmission récessive ont été décrits, ce qui rend parfois le conseil génétique difficile(3,4). L’OI forme un groupe clinique complexe et hétérogène, la classification de Sillence en quatre types étant la plus utilisée(2).
F. H. Glorieux a défini trois autres groupes de patients présentant des particularités cliniques et histologiques distinctes (tableau), ce qui rend la classification plus étendue(1). En dehors de l’atteinte squelettique, d’autres atteintes extrasquelettiques peuvent se voir au cours de l’OI à savoir : une laxité ligamentaire, une coloration bleutée des sclérotiques, une perte de l’audition, des anomalies des tissus vasculaires (dysfonctionnements valvulaires, dilatations, anévrismes ou ruptures des cavités cardiaques, de l’aorte ou des vaisseaux sanguins cérébraux), l’hypercalciurie, liée à l’hyper-remodelage osseux, est aussi habituelle. En cas d’ostéogenèse imparfaite sévère, l’intolérance à la chaleur, la transpiration excessive, l’élévation de la température basale et la tachycardie ont une pathogénie inconnue.
L’impression basilaire se manifeste par des céphalées, une atteinte des derniers nerfs crâniens et une hyper-réflexie. Au cours de l’OI, la fragilité osseuse résulte d’un trouble de la formation osseuse du fait d’une synthèse défectueuse du collagène et d’une perturbation de l’organisation et de l’architecture du tissu osseux(5). Il existe un déséquilibre entre la formation et la résorption en faveur de la résorption osseuse, ce qui justifie l’utilisation des biphosphonates.
Traitement
Le traitement de cette maladie aux multiples facettes est médical, chirurgical et rééducatif. Il n’est pas l’apanage d’une seule spécialité, il doit être interdisciplinaire. Depuis le rapport de Devogelaer en 1987 sur les effets encourageants des bisphosphonates sur un garçon atteint d’ostéogenèse imparfaite, de nombreuses études pilotes tendent à confirmer leur intérêt chez l’enfant souffrant de cette pathologie(6,7).
Les bisphosphonates sont une famille de médicaments qui inhibent puissamment la résorption osseuse en empêchant principalement l’action des ostéoclastes. Les indications actuellement retenues chez l’enfant sont les suivantes : au moins deux fractures annuelles des os longs et apparition de tassements vertébraux. Des nourrissons présentant des formes sévères ont été traités très précocement, avec des résultats très encourageants.
Le biphosphonate le plus utilisé est le pamidronate par voie parentérale mais des formes orales telles que l’olpadronate, l’alendronate (donné à 5 ou 10 mg/j en fonction du poids) et le risédronate ont également fait preuve de leur efficacité(8,9). Le traitement doit être débuté tôt chez les enfants, surtout dans les deux premières années de la vie. Plusieurs effets bénéfiques ont été notés sous bisphosphonates aussi bien sur le plan clinique, densitométrique et histomorphométrique, mais ces derniers ne constituent pas un traitement curatif. Ils doivent s’intégrer dans une prise en charge multidisciplinaire. Le vrai traitement étiologique serait la thérapie génique.
Conclusion
Le pronostic fonctionnel de l’OI dépend de la sévérité de l’atteinte et de sa prise en charge. L’utilisation récente des bisphosphonates, associée à la stimulation motrice et à la chirurgie, a beaucoup amélioré l’autonomie des sujets ayant une forme grave d’ostéogenèse imparfaite. Le pronostic vital est lié à l’atteinte respiratoire corrélée à la sévérité des déformations rachidiennes.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :