Publié le 11 déc 2021Lecture 6 min
Retard de croissance, anémie, douleurs osseuses à 15 ans
Bertrand CHEVALLIER, Boulogne-Billancourt

Sam a 15 ans et 6 mois. Il est adressé en consultation hospitalière pour un retard statural avec un retard pubertaire.
Sa famille
Sam est l’enfant unique d’une famille polonaise arrivée en France trois ans plus tôt. Son père est décédé à 34 ans brutalement, possiblement au décours d’un malaise cardiaque, alors qu’il effectuait des exercices de musculation. Il mesurait 179 cm pour 70 kg, sans antécédents médicaux. Sa mère mesure 161 cm pour 55 kg sans antécédents médicaux. Cette famille est venue rejoindre une partie de sa famille présente depuis dix ans en France.
Sam
Il est né à terme. Sa croissance est régulière jusqu’à 10 ans sur la ligne des -1DS, puis infléchit progressivement sa vitesse de croissance. Il mesure 142 cm pour un poids de 37 kg. Il est scolarisé en 3e avec des difficultés (dyslexie, troubles de l’attention). L’examen clinique note un enfant mince, non dysmorphique. Il est pâle facilement fatigable, peu sportif. Il se plaint de douleurs osseuses diffuses, mais prédominant aux genoux. Il n’a aucun signe pubertaire : les deux testicules bien palpés et la verge sont infantiles ; une discrète pilosité pubienne est présente depuis quelques mois. Il n’y a pas de gynécomastie, pas de taches cutanées. L’abdomen est souple, mais le foie et la rate sont palpés, débordant de 3 cm pour le foie, de 5 cm pour la rate. L’âge osseux est évalué à 12 ans.
Les examens biologiques faits en ville montrent une anémie à 9,7 g/dl ; le reste de la NFS est normal en dehors d’une hypoplaquettose modérée à 98 000/mm. La recherche d’anticorps antitransglutaminases est négative, la VS est à 7 à H1. La testostérone plasmatique est à 0,6 ng/ml, la prolactine et le bilan thyroïdien sont normaux. L’IGF1 est au 25e percentile, les taux de base de la LH et de FSH sont bas.
L’hypothèse d’un retard pubertaire simple ne peut être d’emblée retenue en raison :
d’une inflexion de la vitesse de croissance avant l’âge de 11 ans ;
de la présence d’une anémie dont le mécanisme doit être identifié ;
de la présence d’une hépato-splénomégalie ;
de douleurs osseuses fréquentes, parfois invalidantes
Les explorations vont :
rechercher une anomalie de l’axe hypothalamo-hypophysaire : L’IRM est normale ;
mieux identifier le mécanisme de l’anémie : les réticulocytes sont bas, éliminant une hémolyse. La ferritine est normale, le frottis sanguin n’est pas contributif. Une cause centrale est alors évoquée ;
caractériser l’hépatosplénomeégalie, par une échographie qui précise son caractère homogène, l’absence d’anomalie portale et de masse loco-régionale ;
évaluer l’atteinte osseuse par une scintigraphie osseuse qui note des zones multiples d’hyperfixation au niveau du bassin et des membres et en particulier nette au niveau des deux genoux (figure 1)
Les hypothèses à ce stade privilégient une atteinte de la moelle osseuse primitive ou secondaire : tumorale, surcharge ou constitutionnelle.
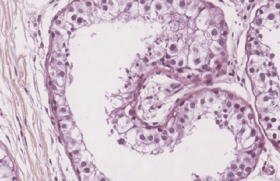
Le myélogramme va suggérer rapidement le diagnostic, identifiant des cellules spécifiques, dites cellules de Gaucher. Un déficit de la glucocérébrosidase (estimée à 15 % de la normale) dosée dans les leucocytes circulants confirme alors le diagnostic de la maladie de Gaucher (figure 2).
La maladie de Gaucher (MG) est une maladie génétique due l’accumulation anormale d’une substance appelée glucocérébroside (autre nom : glucosylcéramide) dans certaines cellules du corps. Cette accumulation entraîne une sensation de fatigue, une augmentation de volume abdominale, des douleurs des os et des saignements du nez, des gencives ainsi que des (hématomes) qui apparaissent en l’absence de traumatisme.
Les manifestations de la maladie de Gaucher sont très variables, ce qui a conduit à classer la maladie en trois types ayant des différences en termes d’âge d’apparition et de gravité.
Le type 1, le plus fréquent (95 cas sur 100), est une maladie de longue durée (chronique). Plusieurs organes sont touchés, essentiellement le foie, la rate et les os. L’âge de début de la maladie et la sévérité des symptômes sont très variables : certaines personnes ont des manifestations très tôt dans l’enfance alors que d’autres n’en auront aucune jusqu’à un âge avancé ou même pendant toute leur vie.
Le type 2, ou type « aigu neurologique » (touchant le système nerveux), très rare (moins de 1 cas sur 100), est le plus sévère. La maladie débute chez le nourrisson de 3 à 6 mois et peut même apparaître avant la naissance (forme fœtale).
Le type 3, ou type « subaigu neurologique », est rare (moins de 5 cas sur 100) et touche l’enfant et l’adolescent. Il existe une atteinte du foie, de la rate, des os et plus rarement du poumon, ainsi que des manifestations neurologiques.
La maladie de Gaucher de type 1 survient plus souvent chez les personnes d’origine juive ashkénaze (jusqu’à 1/1 000 nouveaux cas par an). Elle est généralement due à une anomalie (mutation) du gène GBA, localisé sur le chromosome 1. Le gène code pour la protéine glucocérébrosidase responsable de la dégradation d’un lipide, le glucocérébroside (ou glucosylcéramide), dans le lysosome. Le glucocérébroside est lui-même issu de la dégradation des glycosphingolipides qui sont des constituants des membranes des globules rouges (hématies) et des globules blancs (leucocytes). Dans un fonctionnement normal, ces cellules doivent en effet être détruites lorsqu’elles arrivent en fin de vie, et leurs constituants doivent donc aussi être dégradés. Des mutations du gène GBA (plus de 300 mutations différentes ont été répertoriées) provoquent un déficit en gluco-cérébrosidase (diminution de la qualité et/ou de la quantité de la protéine) plus ou moins important et une accumulation de gluco-cérébroside dans le lysosome. C’est pourquoi la maladie de Gaucher est une maladie de surcharge lysosomale.
La plupart des malades ont moins de 20 ans au moment du diagnostic. Dans le type 1, le foie, la rate, et les os sont les organes les plus fréquemment atteints. Le cœur et les poumons ne le sont que rarement, les reins, la peau, les yeux ou le système digestif ne le sont qu’exceptionnellement. La maladie de Gaucher provoque des atteintes osseuses dans 80 % des cas. Elles sont multiples : des douleurs osseuses (« crises osseuses ») sont fréquentes, souvent intenses, parfois décrites comme une sensation de « broiement » des os. Elles peuvent survenir sur le bassin, les os longs du fémur (os de la cuisse) et des tibias (os de la jambe) et plus rarement sur la tête de l’humérus (os du bras). Ces crises peuvent durer de quelques heures à quelques jours et peuvent nécessiter l’hospitalisation en urgence et la prescription de morphine.
Retard de croissance et retard de puberté
Ces troubles sont fréquents chez les enfants. Il peut également y avoir un retard de la puberté, c’est-à-dire du développement des organes génitaux, du début des règles pour les jeunes filles (qui apparaissent à 16 ans en moyenne), de la pilosité corporelle (pubis et, chez les garçons, barbe et moustache) et de la poussée de croissance associée à cette période. Le retard de puberté se rattrape généralement à la fin de l’adolescence. Comment expliquer les symptômes ? Bien que le déficit enzymatique existe dans toutes les cellules,l’accumulation de glucocérébroside dans les lysosomes affecte principalement un certain type de cellules, les macrophages. Ces macrophages engorgés de lysosomes pleins de glucocérébroside deviennent très volumineux, on les appelle « cellules de Gaucher ». Ces cellules particulières envahissent les organes qui augmentent alors de volume. L’envahissement se fait principalement dans le foie, la rate, lesos et parfois les poumons et d’autres organes. Il est probable que l’infiltration par les cellules de Gaucher comprime les petits vaisseaux qui irriguent ces organes.
Enzymothérapie substitutive
Le traitement enzymatique substitutif (TES ou enzymothérapie substitutive)est utilisé pour traiter les malades atteints de la maladie de Gaucher de type 1. Ce traitement consiste à administrer directement une enzyme identique à la glucocérébrosidase dans le sang du malade. Il permet de rétablir un niveau d’activité enzymatique suffisant pour éliminer en partie la substance accumulée (glucocérébroside) dans les cellules de Gaucher. En 1996,une enzyme de synthèse a été mise sur le marché : l’imiglucérase. Ce traitement doit être poursuivi toute la vie. Il est donné par voie intraveineuse une fois tous les 15 jours. La perfusion dure de 1 à 2 heures. Le traitement a considérablement amélioré l’état des personnes atteintes de la maladie de Gaucher de type 1 (figure 3).
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :