Publié le 11 fév 2008Lecture 9 min
Retard ou absence de puberté chez la fille : quelle attitude adopter ?
D. SAMARA-BOUSTANI, C. DUFLOS-COHADE, E. THIBAUD Unité d’endocrinologie et gynécologie pédiatriques, hôpital Necker Enfants malades, Paris
Quand faut-il s’inquiéter devant un retard pubertaire chez la fille ? Quels sont les signes évocateurs d’un retard d’origine organique ? Quels sont les examens complémentaires permettant d’en déterminer l’étiologie ? Telles sont les questions auxquelles cet article voudrait répondre.
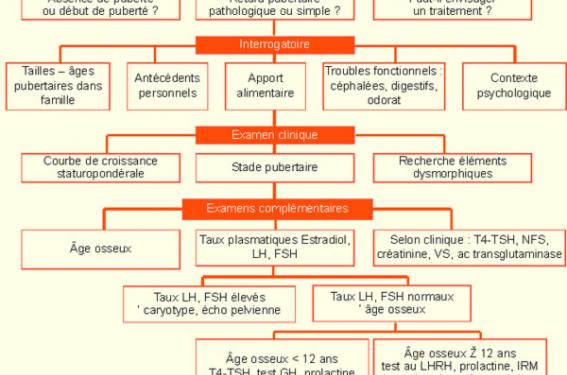
Le retard pubertaire chez la fille se définit par l’absence de développement des seins après l’âge de 13 ans et/ou l’absence de survenue des menstruations après l’âge de 16 ans ou 4 ans après le début du développement mammaire. Il existe donc deux situations : l’impubérisme (absence de développement des seins) et l’aménorrhée primaire (développement des seins présents, mais pas de règles). Impubérisme Devant tout retard pubertaire chez la fille, il faut rechercher des signes en faveur d’une étiologie organique « générale » ou endocrinienne. Le retard pubertaire simple est un diagnostic d’élimination. Il est rare chez la fille. Insuffisances gonadiques (ou hypogonadisme hypergonadotrope) Les insuffisances gonadiques sont responsables d’environ 40 % des retards pubertaires chez la fille. Elles sont congénitales ou acquises. Insuffisances gonadiques congénitales – Syndrome de Turner : étiologie la plus fréquente. Il faut y penser devant un retard de croissance intra-utérin, une petite taille (taille inférieure à -2 DS et/ou un ralentissement de la vitesse de croissance) et des signes dysmorphiques qui peuvent être discrets. Le caryotype est 45 XO ou en mosaïque, ou encore comporte une anomalie de structure d’un chromosome X. – Insuffisance ovarienne à caryotype normal 46 XX. La taille est normale, les ovaires sont de petite taille. Certaines anomalies génétiques ont pu être identifiées : microdélétion du chromosome X, mutation du gène du récepteur de la FSH, mutation du gène Foxl2 (syndrome ptosis-blépharophymosis), prémutation du gène X Fra. Mais dans la majorité des cas, la cause n’est actuellement pas retrouvée. – Dysgénésie gonadique pure 46 XY (phénotype féminin, organes génitaux internes féminins, gonades de petite taille). Les insuffisances gonadiques sont responsables d’environ 40 % des retards pubertaires chez la fille. Insuffisances gonadiques acquises – Secondaire à une radiothérapie : l’insuffisance ovarienne est quasi constante si la dose d’irradiation ovarienne est de plus de 20 grays par irradiation pelvi-abdominale et en cas d’irradiation corporelle totale (conditionnement à une greffe de moelle osseuse). – Secondaire à une chimiothérapie avec des agents alkylants et tout particulièrement après intensification comportant du busulphan ou des fortes doses de cyclophosphamide. – Auto-immune : le plus souvent associée à d’autres atteintes auto-immunes, en particulier dans le cadre de polyendocrinopathies auto-immunes multiples (candidose, hypothyroïdie, hypoparathyroïdie, insuffisance surrénalienne, diabète type 1). – Galactosémie : accumulation intracellulaire du galactose et de ses métabolites. Insuffisance gonadotrope (hypogonadisme hypogonadotrope) Elle peut être permanente ou transitoire. Hypogonadisme hypogonadotrope permanent – Déficit gonadotrope isolé ou associé à des troubles de l’olfaction (anosmie ou hyposmie) dans le cadre d’un syndrome de Kallmann (anomalies de migration des cellules à LHRH et des cellules olfactives). – Syndromes malformatifs : syndrome de Prader-Willi (obésité, retard mental, retard statural, dysmorphie), syndrome de Laurence-Moon-Bardet-Biedl (obésité, retard mental, polydactylie, rétinite pigmentaire). – Insuffisance hypophysaire globale avec déficits somatotrope (GH) et parfois thyréotrope (TSH) ou corticotrope (ACTH) ; elle peut être : – Tumorale : crâniopharyngiome (tumeur la plus fréquente de la région hypothalamo-hypophysaire responsable d’une hypertension intracrânienne, d’un retard statural), germinome, astrocytome et plus rarement adénome à prolactine ; – Congénitale : syndrome d’interruption de la tige pituitaire avec le plus souvent un déficit en GH connu et traité depuis l’enfance ; – séquellaire d’une chirurgie ou d’une radiothérapie (irradiation hypophysaire > 30 Gy) pour tumeur de la région hypothalamo-hypophysaire. Hypogonadisme hypogonadotrope fonctionnel transitoire Toutes les pathologies chroniques avec répercussion nutritionnelle ou syndrome inflammatoire évoluant depuis l’enfance peuvent être la cause d’un retard pubertaire. Mais le retard pubertaire peut parfois révéler la maladie chronique ; c’est le cas en particulier de la maladie de Crohn ou la maladie cœliaque. – L’anorexie mentale Retard pubertaire simple Le retard pubertaire simple reste un diagnostic d’élimination. Il existe souvent un ralentissement statural associé, un retard d’âge osseux et des antécédents de puberté tardive dans la famille. Le retard pubertaire simple reste un diagnostic d’élimination. Conduite à tenir en pratique Le retard pubertaire est attendu Il existe une des pathologies ou un des traitements cités ci-dessus pouvant être responsable d’un hypogonadisme. Le dosage des gonadotrophines FSH et LH de base ou le test au LHRH permettra de confirmer son origine centrale ou périphérique. Le retard pubertaire est le motif de consultation (figure 1) Figure 1. Conduite à tenir devant un retard pubertaire chez la fille. Il faut rechercher : – à l’interrogatoire : des antécédents familiaux de retard pubertaire, un trouble de l’olfaction, des signes digestifs, des troubles du comportement alimentaire ; – à l’examen clinique : un retard staturo-pondéral, des signes généraux (pâleur, adénopathies, etc.) des signes dysmorphiques en faveur d’un syndrome de Turner, des céphalées, des troubles visuels évocateurs d’une hypertension intracrânienne. Les premiers examens à réaliser sont : – un âge osseux, – un dosage de FSH et de LH qui permettra de déterminer si l’hypogonadisme est d’origine périphérique ou centrale. Le taux de FSH et de LH est élevé ; il s’agit d’une insuffisance ovarienne Le caryotype s’impose car la première cause à chercher est le syndrome de Turner (surtout si un retard de croissance est associé). Si le caryotype est normal, il faut rechercher une auto-immunité par la recherche d’atteinte auto-immune associée ; le dosage des anticorps anti-ovariens n’est pas spécifique. Il faut également réaliser une échographie pelvienne pour évaluer la taille des ovaires. Si le bilan auto-immun est négatif, une recherche de cause génétique plus rare est à proposer. Le taux de LH et FSH est normal, il faut alors évaluer l’âge osseux – Si l’âge osseux est ≥ 12 ans, il faut rechercher un déficit gonadotrope par un test au LHRH et éventuellement d’autres déficits hypophysaires associés (GH, TSH et ACTH). Si le déficit gonadotrope est confirmé, une IRM cérébrale doit être réalisée à la recherche d’une tumeur ou d’une anomalie des bulbes olfactifs (en faveur d’un syndrome de Kallmann). – Si l’âge osseux est < 12 ans, le déficit gonadotrope est difficile à diagnostiquer car le test au LHRH est difficilement interprétable et n’a donc pas lieu d’être réalisé. Souvent, ce tableau est associé à un retard statural nécessitant la recherche d’un déficit en hormone de croissance (GH) ou d’une hypothyroïdie. Si ce bilan est normal, le diagnostic le plus probable est un retard pubertaire simple qui sera confirmé par l’évolution clinique. Aménorrhée primaire (Développement normal des caractères sexuels secondaires) Devant toute aménorrhée primaire, il faut en premier lieu rechercher une malformation des organes génitaux par un examen de la vulve et des signes d’hyperandrogénie. L’échographie pelvienne est toujours nécessaire. Le bilan hormonal est orienté par la clinique (figure 2). Figure 2. Conduite à tenir devant une aménorrhée primaire. Devant toute aménorrhée primaire, l’échographie pelvienne est toujours nécessaire. Il n’y a pas de signes d’hyperandrogénie, le développement des caractères sexuels secondaires est normal et complet (stade tanner S5 P5), il faut rechercher une anomalie des organes génitaux : – L’imperforation hyménale est révélée le plus souvent par des douleurs abdominales cycliques et la constitution d’un hématocolpos provoquant un bombement bleuté de l’hymen et une masse pelvi-abdominale palpable parfois par voie abdominale et toujours par le toucher rectal. – Le syndrome de Rokitanski est une aplasie utéro-vaginal. La vulve est normale, mais le vagin est court. L’aplasie utérine est complète, mais peut être partielle. Le diagnostic est clinique et échographique. Il n’y a pas de signes d’hyperandrogénie, le développement des caractères sexuels secondaires est incomplet avec une pilosité pubienne absente ou faible contrastant avec un développement normal des seins (stade tanner S5 P1-2). On suspecte alors une résistance complète aux androgènes : il faut rechercher des gonades en position inguinale, un antécédent de hernie inguinale en période néonatale. L’échographie ne retrouve pas d’utérus et montre des gonades (testicules) en position inguinale. Le caryotype est 46 XY. Le bilan montre un taux de testostérone très élevé, un taux de LH élevé. Il existe une mutation du gène du récepteur aux androgènes, localisée sur le chromosome X. La présence d’une hyperandrogénie clinique (hirsutisme, acné) doit faire rechercher : – Un trouble de l’hormonosynthèse surrénalienne à révélation tardive par : – bloc en 21-hydroxylase (hirsutisme dans 85 % des cas, aménorrhée dans 15 % des cas). Biologiquement, le taux de 17-OHP est élevé surtout après stimulation par le synacthène (ACTH) ; – bloc en 11b-hydroxylase, plus rare et très souvent associé à une hypertension artérielle. Le diagnostic est fait sur l’élévation du composé S de base et après stimulation par le synacthène. – Un syndrome des ovaires polykystiques associant un hirsutisme, souvent une obésité, et parfois un acanthosis nigricans. Les androgènes (testostérone, delta 4-androstènedione) sont élevés. La SHBG (sex hormone binding globulin) est souvent abaissée. Dans un grand nombre de cas, il existe une insulinorésistance avec hyperinsulinémie et parfois intolérance au glucose. À l’échographie pelvienne, les ovaires sont augmentés de volume, globuleux avec un stroma hypertrophié et une échostructure multifolliculaire, mais ces différents critères ne sont pas toujours réunis. Toutes les causes d’hypogonadisme à l’origine d’un retard pubertaire peuvent être responsables d’une aménorrhée primaire lorsque l’hypogonadisme est incomplet ou survient en cours de puberté. Pour la pratique, on retiendra En dehors des situations où le retard pubertaire est attendu, le retard pubertaire chez la fille est à considérer comme pathologique tant que les principales étiologies organiques n’ont pas été éliminées. Il faut orienter le diagnostic par l’interrogatoire, la clinique et le taux des gonadotrophines. Les gonadotrophines sont élevées, il existe donc une insuffisance ovarienne dont la principale cause est le syndrome de Turner. Les gonadotrophines sont normales, il existe un hypogonadisme hypogonadotrope transitoire ou définitif à confirmer par un test au LHRH selon l’âge osseux. Les principales étiologies à rechercher sont la tumeur hypothalamo-hypophysaire et le syndrome de Kallmann (de Morsier) par la réalisation d’une IRM cérébrale. Le bilan est normal, le diagnostic le plus probable est un retard pubertaire simple ou aggravé par des troubles du comportement alimentaire. La conduite à tenir devant une aménorrhée primaire est essentiellement clinique et échographique par la recherche en premier lieu d’anomalie des organes génitaux externes.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :





