Publié le 14 avr 2008Lecture 14 min
Quand et comment diagnostiquer un déficit immunitaire héréditaire ?
C. PICARD*,** *Centre d’étude des déficits immunitaires, Hôpital Necker-Enfants Malades, Paris **Laboratoire de Génétique Humaine des Maladies Infectieuses, Institut National de la Santé et de la Recherche Médicale, U550, Paris
Le diagnostic d’un déficit immunitaire héréditaire (DIH) est important à la fois dans le pronostic et la prise en charge des enfants atteints. Ce diagnostic est clinique avant tout, bien qu’il s’appuie également sur des examens biologiques. Les examens de première intention sont à réaliser en cas d’infections sévères, récurrentes et/ou inhabituelles.
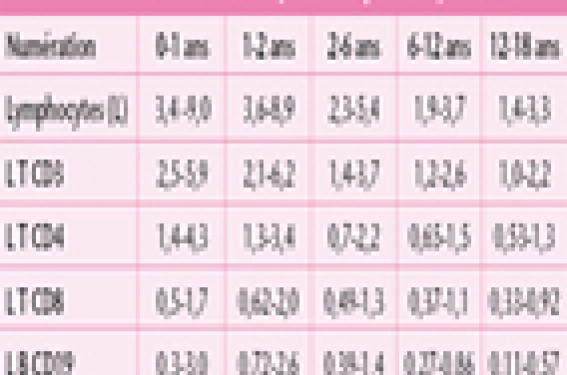
Les examens de première intention sont simples et de routine. Ils permettent d’orienter le diagnostic. L’analyse des antécédents infectieux, l’examen clinique et les résultats de ces examens permettent de guider la prescription des examens de deuxième intention qui dépendront du type de déficit immunitaire héréditaire suspecté. Le but de cet article est de guider le pédiatre dans la démarche diagnostic d’un patient suspecté de déficit immunitaire héréditaire. Les examens de première intention à réaliser lorsqu’un DIH est suspecté Un déficit immunitaire héréditaire doit être recherché devant un ou plusieurs signes cliniques d’alertes (tableau 1) (1). Tableau 1. Signes d’alertes devant faire évoquer un déficit immunitaire héréditaire. • Infections récurrentes pulmonaires et/ou ORL : Fréquence : > 8 otites/an (pendant l’automne et l’hiver) chez les moins de 4 ans ; > 4 otites/an (pendant l’automne et l’hiver) chez les plus de 4 ans ; et plus de 2 pneumonies/an ou > 2 sinusites compliquées/an. • Infections sévères avec des germes encapsulés (pneumocoque, Haemophilus, Neisseria) : un seul épisode de méningite ou sepsis sévère se doit d’être exploré. • Infections à bactéries pyogènes récurrentes (cutanée, invasive, tissulaire, etc.). • Infections récurrentes avec le même type de pathogène. • Infections inhabituelles et/ou d’évolution inhabituelle (ex. infection par un germe opportuniste, diarrhée infectieuse persistante, un muguet ou candidose cutanée récidivante). • Une cassure de la courbe staturo-pondérale et/ou une diarrhée persistante. • Autres signes : eczéma, auto-immunité (ex. cytopénie auto-immune), inflammation chronique ou une lympho-prolifération (adénopathies et hépato-splénomégalie). • Présence d’antécédents familiaux de déficit immunitaire ou de signes cliniques similaires. Les examens à réaliser en première intention sont des examens simples qui vont permettre d’orienter le diagnostic. Ces premiers examens à réaliser sont une numération de la formule sanguine (NFS) plaquettaire, un dosage pondéral des immunoglobulines (Ig) et des sérologies post-vaccinales et/ou post-infectieuse (2). La numération formule sanguine plaquettaire La NFS plaquettaire permet d’apprécier la formule leucocytaire en valeur absolue. Une neutropénie isolée peut être responsable des manifestations infectieuses lorsqu’elle est généralement inférieure à 500/mm3. Une numération normale des polynucléaires neutrophiles sur un examen n’élimine pas une neutropénie cyclique, celle-ci devra être recherchée par des NFS successives hebdomadaires. Les lymphocytes chez le jeune enfant, doivent être impérativement interprétés en fonction des normes pédiatriques (tableau 2)(3). Une lymphopénie orientera vers un déficit de l’immunité cellulaire (immunité dépendante des lymphocytes T). La NFS peut apporter d’autres informations, telle qu’une anémie, une thrombopénie et/ou une neutropénie dans un contexte de déficit de l’immunité cellulaire, qui peuvent être des manifestations d’auto-immunité, complication fréquente de ce type de déficit immunitaire. L’analyse du frottis sanguin permet de rechercher des corps de Jolly lorsqu’une asplénie est suspectée. Une neutropénie cyclique devra être recherchée par des NFS successives hebdomadaires. Le dosage pondéral des immunoglobulines sériques Le dosage pondéral des immunoglobulines apporte des éléments aux diagnostics des déficits immunitaires humoraux (lymphocytes B) et des déficits immunitaires combinés (touchant à la fois les lymphocytes T et B). Ce dosage pondéral des IgG, des IgA et des IgM doit être interprété en fonction de l’âge chez les enfants ; en effet il existe des variations importantes des taux au cours de l’enfance (tableau 3)(2). Il est également important de savoir que le taux d’IgG est difficilement interprétable avant l’âge de 4 mois, car à cet âge l’essentiel des IgG sont d’origine maternelle. En cas d’anomalies des explorations de premières intentions, un dosage des sous-classes des IgG (1, 2, 3 et 4) sera proposé aux enfants de plus de 20 mois, cet examen étant difficilement interprétable avant cet âge. Ces dosages d’Ig permettent d’apprécier la production globale d’anticorps sans tenir compte de leur spécificité. Le taux d’IgG est difficilement interprétable avant l’âge de 4 mois. Les sérologies post-vaccinales et post-infectieuses L’étude des sérologies post-vaccinales après vaccination et des sérologies après une infection permet d’apprécier la capacité de production d’anticorps spécifiques qui peuvent être de deux types, soit de type antiprotidique, soit de type antipolysaccharidique. • Les anticorps de type antiprotidique font appel à une coopération entre les lymphocytes T et B. Ils sont produits au décours d’une infection ou après une vaccination conjugué (par exemple : vaccin antidiphtérique, vaccin conjuguée anti-Haemophilus). • Les anticorps de type antipolysaccharidique font eux appel à une réponse lymphocytaire B seule. Ils sont produits après une infection par un germe encapsulé (par exemple : Streptococcus pneumoniae) ou bien après une vaccination par les vaccins non conjugués antipneumocoque ou antiméningocoque. Les allohémagglutinines de groupe sanguin sont également des anticorps de type antipolysaccharidique ; ce sont des IgM naturelles dirigées contre les antigènes des groupes sanguins A et/ou B. L’enfant de moins de 2 ans présente de manière physiologique un défaut de production de ces anticorps antipolysaccharidiques : la production de ces anticorps n’est donc pas évaluable avant l’âge de 2 ans. L’ensemble des sérologies doit être interprété avec prudence pendant les 6 premiers mois de vie, période pendant laquelle il peut exister des sérologies faussement positives dues à la persistance d’IgG maternelles chez l’enfant. Examens biologiques de deuxième intention Dans la grande majorité des cas, l’ensemble des éléments apportés par les examens simples conjointement à ceux apportés par l’anamnèse et l’examen clinique permet de cibler les examens à demander en deuxième intention. Le phénotypage des lymphocytes Cet examen quantitatif permet par l’intermédiaire de molécules membranaires exprimées de caractériser les populations lymphocytaires. La molécule CD3 est spécifique des lymphocytes T, qui se repartissent en deux sous-populations CD4 et CD8. Les molécules CD19 et CD20 sont spécifiques des lymphocytes B. Les cellules Natural Killer (NK) expriment les molécules CD56 et CD16. La numération des lymphocytes T, des sous-populations lymphocytaires T, des lymphocytes B et NK doit être interprétée en valeur absolue et en fonction de l’âge du patient, car il existe des variations physiologiques importantes pendant les premières années de vie (tableau 2) (3). Ces marqueurs sont utilisés le plus fréquemment, mais d’autres peuvent être utilisés lorsqu’un phénotypage des populations lymphocytaires plus poussé est nécessaire. La numération des lymphocytes T, des lymphocytes B et NK doit être interprétée en valeur absolue et en fonction de l’âge du patient. Études fonctionnelles des lymphocytes T La fonction des lymphocytes T peut être évaluée in vitro par le test de transformation lymphoblastique (TTL) ou test de prolifération lymphocytaire T. Ce test mesure la capacité proliférative des lymphocytes T vis-à-vis de différents facteurs qui peuvent être soit des mitogènes, soit des antigènes. Les mitogènes (ex. la PHA) sont capables de stimuler les lymphocytes T de manière non spécifique, c’est-à-dire sans immuni-sation préalable du patient. Une sensibilisation antérieure du patient est nécessaire pour explorer la réponse aux antigènes, soit par infection (par ex. candidose), soit par vac- cination (par ex. anatoxine tétanique, tuberculine). Cette réponse n’est interprétable que durant l’année qui suit la vaccination. Un test négatif au-delà de ce délai nécessite une confirmation après un rappel vaccinal (2). Études fonctionnelles des polynucléaires neutrophiles Les polynucléaires neutrophiles ont plusieurs fonctions : le mouvement, l’adhésion, l’endocytose et la destruction des agents pathogènes ingérés. Le mouvement et l’adhésion des polynucléaires sont explorés par l’étude du chimiotactisme spontané et en présence de substances chimio-attractantes. Les fonctions phagocytaires qui étudient l’explosion oxydative (mécanisme mis en jeu pour détruire le pathogène phagocyté) font appel à la capacité de réduction, grâce à la génération entre autres d’ions superoxyde O2-. Le test de réduction au nitro bleu de tétrazolium (NBT) permet de détecter cette réduction en microscopie optique dans les polynucléaires neutrophiles. Cette capacité de réduction des leucocytes peut être évaluée par cytométrie de flux ou par chimioluminescence(4). Ces trois tests explorent de manières différentes, mais complémentaires l’explosion oxydative des leucocytes. Explorations du complément La voie classique du complément est explorée par le dosage du CH50. Un défaut d’un des composés du Cl au C9 entraîne une baisse du CH50, et le diagnostic sera confirmé par le dosage spécifique des divers composants(5). La voie alterne du complément est étudiée par le dosage de l’AP50 ; en cas d’anomalie, l’exploration sera complétée par le dosage des différents composés (D, H, I et properdine). Des défauts génétiques de tous les composés ont actuellement été décrits. Parmi les déficits du complément, les déficits en C3, C4, C5, C6, C7, C8 et en properdine sont plus particulièrement responsables d’infections bactériennes. Les déficits en protéines H et I, entraînant un déficit en C3 par consommation, peuvent être responsables d’infections bactériennes récurrentes. Quand et commentpratiquer ces explorations de 2e intention ? Les examens de 2e intention sont à pratiquer devant un ou des signes d’alerte (tableau 1) et après les examens de 1re intention dans les situations suivantes : En cas d’anomalie des examens de 1re intention (figure 1) : Figure 1. Anomalie des examens de 1re intention : algorithme de prescription des examens de 2e intention. Si les examens de 1re intention mettent en évidence un défaut production d’anticorps post-vaccinaux isolé. Il faut revacciner l’enfant, puis contrôler le taux d’anticorps 3 à 6 semaines après cette nouvelle vaccination. Si le taux d’anticorps reste bas, il faut compléter le bilan par un phénotypage lymphocytaire et des TTL. En présence d’une lympho-pénie isolée à la NFS, il faut la contrôler quelques jours plus tard pour vérifier sa normalisation. Si elle persiste, il faut compléter le bilan par un phénotypage lymphocytaire et des TTL. En présence d’un déficit en IgA isolé, qui est rarement symptomatique, mais fréquent (1 cas sur 600 dans la population générale), il faut éliminer un déficit immunitaire de l’immunité cellulaire associé (phénotypage lymphocytaire et des TTL) ou un déficit en sous-classes IgG2 et IgG4 associés si l’enfant est symptomatique. En cas d’hypogamma-globulinémie et de sérologies anormales, il faut réaliser un phénotypage lymphocytaire et des TTL : – si ces examens sont normaux, un phénotypage lymphocytaire B plus détaillé et des études fonctionnelles B seront à réaliser pour mieux caractériser ce déficit de l’immunité humorale isolé ; – si le taux d’IgM sérique est normal ou élevé, contrastant avec des taux d’IgG et d’IgA effondrés, le diagnostic de syndrome hyper-IgM sera évoqué. Il existe dans ce cas une anomalie de commutation isotypique et des explorations supplémentaires pour caractériser ce syndrome hyper-IgM seront à réaliser ; – si le phénotypage lymphocytaire B est anormal, par exemple les lymphocytes B sont absents de façon isolée, le diagnostic évoqué sera l’agammaglobulinémie. Cette maladie est généralement liée à l’X (agammaglobulinémie de Bruton), mais il existe également de rares formes autosomiques récessives ; – si le phénotypage lymphocytaire T et les TTL sont anormaux, le diagnostic de déficit immunitaire combiné sera évoqué (cf. point suivant). Si l’enfant présente une lymphopénie associée à une hypogammaglobulinémie et à des sérologies anormales. Le bilan sera complété par un phénotypage lymphocytaire et des TTL. – en cas d’absence de lymphocytes T (ou la présence d’une lymphopénie T profonde), l’enfant présente un déficit immunitaire combiné sévère (DICS). Ces enfants avec un DICS présentent des infections récurrentes à tout type de pathogènes, en particulier des infections opportunistes et virales, dès les premières semaines de vie ; – lorsque la lymphopénie T est moins profonde avec des TTL anormaux, on parle de déficit immunitaire combiné (DIC). Les DIC associent un défaut de l’immunité cellulaire (immunité dépendante des lymphocytes T) à un défaut de l’immunité humorale (immunité dépendante des lymphocytes B). Les patients avec des DIC se révèlent plus tard dans la vie que les enfants atteints de DICS. Les causes génétiques du DIC seront recherchées en fonction du tableau clinique, de la transmission génétique du déficit et des résultats des explorations immunologiques. En cas de normalité des examens de 1re ligne (figure 2) : Figure 2. Normalité des examens de 1re intention : algorithme de prescription des examens de 2e intention. Si le patient présente des infections bactériennes systémiques sévères causées par des germes encapsulés (ex. pneumocoque). Il faut rechercher des corps de Jolly au frottis sanguin à la recherche d’une asplénie ou d’une hyposplénie, et explorer le complément par les dosages du CH50 et de l’AP50. Un défaut d’une des sous-unités du complexe d’attaque membranaire du complément (C5, C6, C7, C8 ou C9) ou de la properdine (voie alterne) entraîne des infections à Neisseiria. Les défauts en C3, C4, facteurs H et I entraînent des infections bactériennes récurrentes. En cas de normalité de ces explorations, il faut rechercher des arguments cliniques et biologiques pour les défauts génétiques de l’immunité innée (défaut complet récessif d’IRAK4 et le déficit en NEMO)(6). Le déficit en NEMO associe généralement au déficit immunitaire une dysplasie ectodermale anhydrotique. Si le patient présente des infections sévères tissulaires bactériennes et/ou fongiques (ex. abcès cutanés et/ou viscéraux, pneumopathie). Il faut réaliser des études fonctionnelles des du chimiotactisme des polynucléaires, le diagnostic de défaut des molécules d’adhésion leucocytaire sera évoqué. Le diagnostic de granulomatose septique chronique sera évoqué en cas d’explosion oxydative leucocytaire anormale. Si ces deux premières explorations sont normales, un dosage des IgE sera à réaliser pour rechercher des arguments biologiques en faveur d’un syndrome hyper-IgE (ou syndrome de Job ou Buckley). En cas d’infections mycobatériennes causées par des mycobactéries environnementales ou bien par le bacille vaccinal de Calmette et Guérin (BCG). Le syndrome de prédisposition mendélienne aux infections mycobactériennes sera suspecté(6). C’est un syndrome hétérogène se caractérisant par la survenue d’infections mycobactériennes et chez la moitié des patients des infections par des salmonelles ont également été observées. Des mutations causales dans cinq gènes autosomaux intervenant dans la réponse ou la production de l’interféron gamma ont été décrites chez des patients avec ce syndrome. Conclusion En cas de normalité de l’ensemble de ces explorations immunitaires, il ne faut cependant pas éliminer le diagnostic de déficit immu-nitaire héréditaire, car c’est avant tout un diagnostic clinique et les explorations réalisées actuellement n’explorent qu’une partie limitée du système immunitaire. De plus, l’observation clinique attentive permet le diagnostic de nouveaux déficits immunitaires, ainsi que le développement de nouvelles explorations immunitaires. En effet, depuis une dizaine d’années des infections sévères qui étaient considérées comme idiopathiques ont maintenant une explication génétique(6). En cas de suspicion de déficit immunitaire héréditaire, il ne faut pas hésitez à demander un avis auprès d’un immuno-logiste pédiatre pour orienter au mieux l’exploration du patient. Apres avoir porté le diagnostic de déficit immunitaire, il faudra evaluer le retentissement de celui-ci.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :