Rhumatologie - Os - Orthopédie - Traumatologie
Publié le 04 sep 2006Lecture 19 min
Maladies monogéniques en rhumatologie
D. Floret - Hôpital édouard-Herriot, Lyon
Si l’épiglotitte aiguë est devenue rare, elle a gardé toute sa gravité. Bien que l’étiologie bactérienne ait changé, du fait de la quasi-disparition Hæmophilus influenzæ, les principes thérapeutiques restent les mêmes et reposent sur la sécurisation des voies aériennes supérieures par l’intubation et sur une antibiothérapie à large spectre. L’enfant intubé est pratiquement guéri, encore faut-il que le geste ait été réalisé à temps et dans de bonnes conditions de sécurité.
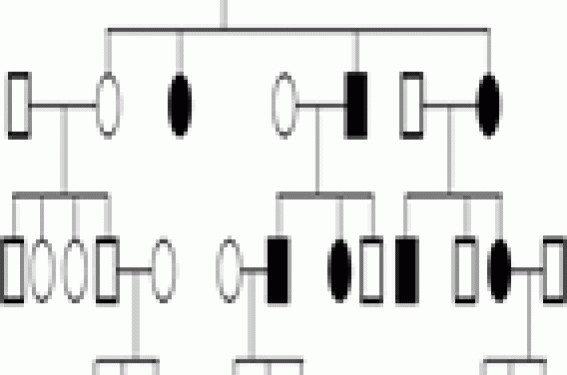
Une maladie monogénique est une maladie génétiquement déterminée, due à un défaut dans un seul gène et obéissant à une hérédité mendélienne ou mitochondriale. Une telle maladie n’est pourtant pas toujours transmise par l’un des 2 parents. Il existe en effet des accidents de novo (exemple : achondroplasie avec 80 % de néomutation dans le gène FGFR3). Les maladies monogéniques sont déterminées par la présence d’un ou des 2 allèle(s) mutant(s) sur la même paire chromosomique, à un locus donné. Lorsqu’une personne possède une paire d’allèles identiques, elle est homozygote pour le caractère considéré ; lorsque les allèles sont différents, elle est hétérozygote. Les maladies monogéniques sont le plus souvent des maladies rares et prises en charge principalement par les pédiatres et les généticiens médicaux. Grâce aux progrès récents, certains patients atteints de maladies génétiques ont une espérance de vie qui s'allonge. Les médecins d’adulte sont donc de plus en plus souvent conduits à les prendre en charge. Ils peuvent aussi être amenés à poser un diagnostic, si ceci n’a pas été fait dans l’enfance. Le but de cette revue est d’abord de proposer des bases permettant d’évoquer le diagnostic de maladie génétique, puis certains éléments pour tenter de le préciser. Nous avons ensuite décrit certaines de ces pathologies parmi les plus fréquentes ou celles dont le diagnostic est devenu indispensable du fait des progrès thérapeutiques. Rappels 1 Hérédité autosomique dominante (figure 1) Figure 1.- Hérédité autosomique dominante : transmission verticale. Le phénotype s'exprime à l’état hétérozygote même si un seul des deux allèles d’un même gène porte une mutation. Chaque individu atteint dans un arbre généalogique a un parent atteint (sauf s’il existe une néomutation) et ainsi de suite, révélant une transmission verticale. La transmission peut se faire de père en fils contrairement à l'hérédité dominante liée au chromosome X : par exemple la neurofibromatose de von Recklinghausen et l’achondroplasie. La pénétrance et l’expressivité sont deux caractéristiques de la dominance. La pénétrance est la probabilité qu’un gène puisse avoir une expression phénotypique (pourcentage des porteurs de la mutation qui ont le phénotype). L’expressivité est le degré d’expression du phénotype (variation des manifestations d’un patient à l’autre et même parfois au niveau intrafamilial). Hérédité autosomique récessive, transmission horizontale (figure 2) Figure 2.- Hérédité autosomique récessive : transmission horizontale. Le phénotype s’exprime uniquement si les deux allèles portent une mutation (homozygotes versus hétérozygotes composites). Il est important de rechercher une consanguinité et de préciser l’origine ethnique, comme dans la maladie de Gaucher. Hérédité liée au chromosome X (figure 3) Figure 3.- Hérédité liée au chromosome X. Il est désormais acquis que les femmes peuvent être atteintes du fait de l’inactivation aléatoire d’un X. Elle présentent alors des formes moins sévères que les hommes (d’où des diagnostics parfois tardifs). Les femmes sont généralement conductrices et les hommes atteints. Toutes les filles et aucun des garçons d’un individu de sexe masculin atteint sont affectées. Les femmes hétérozygotes sont habituellement saines, mais certaines peuvent exprimer la maladie avec une sévérité variable (processus de lyonisation = inactivation aléatoire d’un X), comme dans le cas de l’hémophilie A, la dystrophie musculaire de Duchenne et le rachitisme hypophosphatémique. Hérédité mitochondriale L’hérédité se fait uniquement à travers les mères par les mitochondries de l’ovocyte. Un homme peut être atteint de la maladie, mais aucun homme ne peut la transmettre. Mosaïcisme La mutation ne concerne pas les gamètes, mais se produit plus tard, après la fécondation (accident postzygotique) au cours des divisions cellulaires survenant pendant l’embryogenèse. L’individu est alors constitué de deux lignées cellulaires dérivant d’un même zygote, dont l’une est porteuse de la mutation. Un exemple est la dysplasie fibreuse, dont la mutation, si elle affecte le gamète, n’est pas compatible avec la vie. Seul le mosaïcisme est viable, et l’atteinte sera plus ou moins étendue, concernera le squelette seul ou associera des lésions osseuses, cutanées et endocriniennes (syndrome de McCune-Albright) selon le stade de développement embryonnaire plus ou moins tardif auquel survient la mutation. Il existe d’autres modes d’hérédité non traditionnelle que nous ne développerons pas ici : l’empreinte génomique parentale, la disomie uniparentale et le digénisme. Comment évoquer et établir un diagnostic de maladie génétique ? Histoire familiale Les maladies génétiques se caractérisent par le risque d’atteinte familiale. Pour établir le mode de transmission, la première étape consiste à obtenir l’histoire familiale du patient et d’en résumer les points importants sous la forme d’un arbre généalogique. Il conviendra de rechercher des formes d’expression mineure de la maladie. Le mode de transmission dépend essentiellement de deux facteurs : • la localisation chromosomique du gène, qui peut être autosomique ou liée au chromosome X ; • l’expression dominante ou récessive du phénotype. Examen clinique L’examen clinique devra être descriptif, complet, rigoureux et méthodique. Les critères morphologiques mineurs représentent des éléments essentiels pour poser certains diagnostics. L’existence d’un retard mental doit être recherché. La gravité du syndrome repose sur l’existence d’une pathologie d’organe ou multisystémique, mais c’est parfois l’aspect facial qui permet une définition du syndrome et une reconnaissance de la pathologie. On analysera la morphologie cranio-faciale, les phanères et les extrémités. L'étude de la morphologie cranio-faciale (figure 4) comprend l'analyse de la forme du crâne et ses dimensions, la forme du visage et l'analyse des différents étages de la face. Figure 4.- Patiente atteinte d’une microdélétion 22q11.2. La dysmorphie faciale est évocatrice du diagnostic. On retrouve une microrétrognatie, une pointe de nez bulbaire et une hypoplasie des ailes du nez. On peut noter, par exemple, la présence d’un hyper ou d’un hypotélorisme (distance interpupillaire), l’existence d’une protusion ou d’une rétraction (le front, le philtrum et le menton sont normalement sur la même ligne verticale), l'existence d’une rétrognathie, d’une prognathie ou d’une micrognathie. L'examen des phanères comprend entre-autre, l'étude de l'implantation des cheveux, et d'un défaut ou d'un excès de pilosité. L'examen des extrémités consiste en l'étude de la longueur relative des doigts, le contour des muscles intrinsèques, la recherche de syndactylies cutanées, de contracture ou de laxité articulaire et la mesure globale ainsi que celle des différents segments. Données radiologiques Les examens radiologiques à réaliser seront guidés par l’interrogatoire et l’examen clinique. Le plus souvent, on réalisera des radiographies du crâne, du rachis, du bassin, des os longs et dans certains cas des extrémités. L’examen sera bilatéral et comparatif. Les anomalies observées permettront de classer la pathologie recherchée : ostéochondrodysplasie ou dysostose (figure 5). Figure 5.- Maladie des synostoses multiples. Fusion des 2e et 3e phalanges ainsi que des os du carpe. On recherchera des anomalies de la croissance (au niveau du rachis, du bassin, des épiphyses ou des métaphyses des os longs), de la structure de l’os (anomalies du modelage ou de la densité osseuse) ou du cartilage (développement anarchique de tissu cartilagineux ou du tissu fibreux). Confirmation du diagnostic Si l’ensemble des données cliniques et radiographiques peut permettre d’évoquer une maladie génétique, le diagnostic exact et sa confirmation nécessiteront souvent une discussion pluridisciplinaire (radiologue, généticien), des dosages biologiques (enzymatique) et des analyses génétiques (caryotype classique ou FISH, génétique moléculaire). L’annonce du diagnostic nécessite une connaissance approfondie de la pathologie et sera le plus souvent réalisée en consultation de génétique médicale. Les diagnostics à connaître Dans leur pratique, les rhumatologues pourront être confrontés à certaines maladies génétiques. Quelques unes d’entre elles sont répertoriées ici, celles dont la fréquence ou les possibilités thérapeutiques nouvelles nécessitent le diagnostic. Hémochromatose génétique liée à HFE-12 Cette forme représente la grande majorité des hémochromatoses de l’adulte. Il s’agit d’une des seules maladies génétiques qui n’est pas une maladie rare, avec une prévalence de 1/1000. La pathologie est de transmission autosomique récessive. Elle est le plus souvent provoquée par une mutation C282Y du gène HFE-1 sur le bras court du chromosome 6. Les patients doivent être homozygotes pour cette mutation ou hétérozygotes composites avec une mutation H63D ou S65C sur l’autre allèle. L’asthénie et les manifestations articulaires sont deux des signes les plus précoces. On recherchera une mélanodermie, une hépatomégalie, des perturbations du bilan biologique (hyperferritinémie, augmentation du coefficient de saturation de la transferrine et perturbation du bilan hépatique) et des atteintes endocriniennes (diabète et hypogonadisme). Les rhumatologues évoqueront le diagnostic devant des arthralgies, principalement de l'articulation radio-ulnaire et des 2e et 3e articulations métacarpophalangiennes (MCP). Sur le plan radiographique, on retiendra des lésions de chondrocalcinose articulaire, un pincement de l’interligne et une ostéophytose en « hameçon » au niveau des articulations MCP (figure 6). Sur le plan biologique, il existe une élévation du taux de saturation de la transferrine (> 45 %). Un test génétique simple permet la confirmation du diagnostic. Figure 6- Hémochromatose. Ostéophytose caractéristique des métacarpophalangiennes avec aspect dit en « hameçon » ou en « crochet ». Les anomalies de développement du squelette (ostéochondrodysplasies) Le diagnostic est le plus souvent porté dans l’enfance ou l’adolescence. Il est impossible de décrire ici l’ensemble de ces pathologies. Le clinicien devra déterminer le mode de transmission en reconstituant l’arbre généalogique – tout en sachant que de nombreux cas sont sporadiques – regrouper l’ensemble des anomalies constatées et réaliser une étude radiologique. Ces éléments permettront de distinguer les atteintes épiphysaires, métaphysaires, diaphysaires et rachidiennes. On distinguera aussi les exostoses multiples, les enchondromatoses, les dysplasies fibreuses, mieux connues des rhumatologues, les anomalies de la densité de la corticale diaphysaire des os longs et du modelage. Cette individualisation permet ensuite de se référer à l’ouvrage de Pierre Maroteaux et de Martine Le Merrer3 pour retenir un diagnostic précis à confirmer avec l’aide d’un généticien. Citons toutefois : > La dysplasie spondyloépiphysaire tardive4 qui associe une insuffisance staturale (particulièrement au niveau du tronc) sans dysmorphie faciale et une arthrose précoce. L’examen radiologique retrouve des vertèbres ovoïdes évoluant vers un aplatissement généralisé (platyspondylies nettes dans la région lombaire) et une atteinte épiphysaire de l’extrémité supérieure du fémur. La transmission est récessive, liée au chromosome X et touche exclusivement les garçons. La distinction avec d’autres dysplasies spondylo-épiphysaires (dont certaines maladies métaboliques : syndrome de Morquio, mucolipidose de type III, etc.) est indispensable du fait du pronostic et du mode de transmission différents. > La dysplasie fibreuse est provoquée par une mutation somatique dominante en mosaïque, activatrice (gain de fonction) du gène codant pour la protéine Gs 5. Son association avec une endocrinopathie (le plus souvent une puberté précoce) et des taches café au lait sur la peau réalise le syndrome de McCune-Albright. La consultation peut être motivée par la découverte d’une asymétrie corporelle. L’examen radiologique retrouve des zones claires arrondies ou polycycliques évocatrices du diagnostic. > L’ostéogenèse imparfaite6 se caractérise par sa grande hétérogénéité phénotypique (4 types selon la gravité) et génotypique (mutation COL1A1 et COL1A2). Le diagnostic sera évoqué devant des fractures à répétition apparaissant à l'occasion de traumatismes minimes pendant l'enfance ou devant une transparence excessive du squelette. La présence de sclérotiques bleues et d’une surdité de transmission sont des arguments supplémentaires. Le plus souvent la transmission est autosomique dominante, mais le conseil génétique devra être très prudent devant le risque non négligeable de mosaïque germinale et de mosaïque somatique chez un des deux parents. > L’ostéopétrose7, et plus particulièrement la maladie d’Albers-Schönberg, dont le diagnostic peut être tardif. La découverte peut être fortuite, suite à une fracture ou à des douleurs ostéo-articulaires (épiphysiolyse, luxation post-traumatique, arthrite septique). Sur le plan radiologique, on retrouvera une densification généralisée et des régions métaphysaires mal modelées et élargies. La transmission est peut-être autosomique dominante (gène CLCN7) ou autosomique récessive (gène CAII, TCIRG1, CLCN7 et gl/gl). > La maladie de Camurati-Engelmann qui peut provoquer des troubles de la marche et des douleurs des jambes. Il s’agit d’une maladie autosomique dominante avec une expressivité variable. Ce syndrome est lié à des mutations du gène TGFB1 (Transforming Growth Factor Beta 1). Le diagnostic sera évoqué devant l’aspect fusiforme de la corticale diaphysaire des os longs (souvent bilatérales et symétriques) (figure 7). Figure 7- Maladie de Camurati-Engelman : dysplasie diaphysaire responsable de l’aspect fusiforme de la corticale diaphysaire. > La chondrodysplasie métaphysaire dominante de type Schmid qui associe une petite taille sans dysmorphie du visage et une coxa vara. Maladies inflammatoires Ce groupe de maladies, à type de fièvre périodique avec ou sans signes articulaires, a largement bénéficié des progrès récents en génétique moléculaire notamment pour le diagnostic. La fièvre méditerranéenne familiale Elle est liée à une mutation du gène MEFV sur le bras court du chromosome 16, touche la population du bassin méditerranéen et s'exprime par des crises fébriles brèves à intervalles variables, des douleurs intermittentes de l'abdomen, du thorax, et des articulations et associe des manifestations cutanées. Elle peut se compliquer d'une amylose. La transmission est autosomique récessive. Le gène en cause code pour une protéine appelée marenostrine exprimée dans les granulocytes, les monocytes et les éosinophiles. Le diagnostic peut être confirmé par l’étude moléculaire dans plusieurs laboratoires en France. Le syndrome périodique ou TRAPS Il est dû à une mutation du gène TNFR1 ou TNFRSF1A de transmission autosomique dominante. Les manifestations cliniques sont : des épisodes paroxystiques caractérisés par une forte fièvre avec frissons durant 2 à 3 semaines, des douleurs abdominales diffuses avec nausées et vomissements, des symptômes d'appendicite, une pseudocellulite et des myalgies. Le syndrome de fièvre avec hyperIgD Il se traduit par des accès de fièvre périodique qui surviennent toutes les 4 à 8 semaines avec une réaction inflammatoire intense, accompagnée d'adénopathies, de douleurs abdominales, de diarrhées, de douleurs articulaires, d'hépatosplénomégalie, et de signes cutanés. Le gène responsable MVK code pour une enzyme : la mévalonate kinase. Il est situé en 12 q24 et se transmet de manière autosomique récessive. Le diagnostic repose sur la mise en évidence du déficit enzymatique révélé soit par une mévalonaturie pendant les accès fébriles, soit par dosage de la mévalonate kinase lymphocytaire pendant ou en dehors des accès, ainsi que par une augmentation des IgD. Parmi les maladies de type fièvre périodique, citons aussi le CAPS (Cryopyrine associated periodic syndrome) qui est lié au gène CIAS1 (code-induced auto-inflammatory syndrome 1) et qui regroupe trois entités phénotypiques : l’urticaire au froid familial, le syndrome de Muckle-Wells et le syndrome CINC. Maladies métaboliques Homocystinurie C’est une maladie de transmission autosomique récessive due à un déficit en cystathionine ß-synthase. Ce tableau classique associe un retard mental, des complications thromboemboliques et un aspect marfanoïde et un risque de luxation articulaire ou de sub-luxation du cristallin. En l’absence de traitement (pyridoxine), une ostéoporose précoce peut apparaître. Maladie de Gaucher10 Cette maladie est due au déficit de la glucocérébrosidase provoquant l’accumulation du glucocérébroside dans les cellules du système réticulo-endothéliale, principalement dans les macrophages. Le mode de transmission est autosomique récessif. La fréquence est de 1/60 000 à 1/100 000 chez les caucasiens, mais beaucoup plus fréquente dans la population juive ashkénaze (incidence = 1/500). Les rhumatologues pourront être confrontés au type 1 qui épargne le système nerveux central. Il se caractérise par l’existence d’une hépatosplénomégalie, d’une pancytopénie, et des signes osseux à type d’ostéopénie, de déformations plus ou moins caractéristiques (élargissement en flacon d’Erlenmeyer), d’ostéonécrose, de douleurs et de fractures. La très grande hétérogénéité clinique explique la fréquence des diagnostics tardifs. Le diagnostic sera établi suite au dosage enzymatique. Maladie de Hurler (mucopolysaccharidose de type I10) Cette maladie de surcharge est provoquée par un déficit en alpha-L-iduronidase. Le mode de transmission est autosomique récessif. Les rhumatologues peuvent être confrontés à certaines formes modérées chez l’adulte (type Scheie) diagnostiquées tardivement. L’examen clinique retrouvera : – des déformations faciales avec une ensellure nasale, des narines larges, des lèvres et des oreilles épaisses, des cheveux hirsutes et de gros calibre et un front proéminent ; – un tronc court déformé par une cyphose dorsale basse ou lombaire ; – des membres trapus et légèrement en flexion. Les amplitudes articulaires sont diminuées et, progressivement, on observe des doigts fléchis en griffe irréductible. Il s’y associe une hépatosplénomégalie. Sur le plan radiologique, l’hypoplasie antérieure d’une vertèbre lombaire avec une cyphose (figure 8) peut être vue précocement. Il existe un élargissement de la diaphyse des os longs et fréquemment une dysplasie épiphysaire. Les métacarpiens sont coniques et mal modelés. (A : aspect radiologique et B : aspect IRM) du corps vertébral de L2. Maladie de Fabry Il s’agit d’une maladie métabolique, de transmission liée au chromosome X, due au déficit en alpha-galactosidase A responsable de l’accumulation de globotriaosylcéramide. Des crises douloureuses aigues paroxystiques de la paume des mains et de la plante des pieds mais aussi des acroparesthésies chroniques pourront motiver une consultation auprès d’un rhumatologue. L’association d'angiokératomes, d'une hypohidrose, de dépôts cornéens, d'une atteinte rénale, de manifestations cardio-vasculaires et/ou de manifestations neurologiques permettront d'évoquer le diagnostic. Un dosage enzymatique est nécessaire pour la confirmation du diagnostic. En raison du phénomène de lyonisation, les femmes hétérozygotes peuvent présenter des symptômes de la maladie. Autres maladies Citons aussi le syndrome de Gitelman (pathologie de transmission autosomique récessive, caractérisée par une alcalose hypokaliémique, une hypomagnésémie et une hypocalciurie) qui s’associe souvent à une chondrocalcinose, la phénylcétonurie dont le diagnostic est réalisé lors du dépistage néonatal systématique en France, qui peut évoluer vers une ostéopénie, et l’ensemble des maladies lysosomales (maladie de Hunter, syndrome de Morquio, mucolipidose, etc.) dont l’évocation doit motiver un dosage biochimique des enzymes lysosomales. Maladie de Paget Il s’agit d’une pathologie bien connue des rhumatologues pour laquelle le diagnostic ne pose aucun problème. Récemment, une mutation dans le gène SQSTM1 codant pour le sequestosome 1 (p62) a été identifiée comme une cause de forme familiale et même de certains cas sporadiques. Nous ne parlerons pas ici des maladies neurogénétiques qui peuvent être orientées vers une consultation de rhumatologie, ni des déficits immunitaires qui peuvent favoriser des troubles autoimmuns. Conclusion L’utilisation de la méthode décrite dans le premier chapitre, ainsi que de multiples contacts avec nos collègues radiologues et généticiens nous ont permis d’identifier 54 patients atteints de maladie monogénique et admis pour des symptômes osseux ou articulaires dans le service de rhumatologie du CHU de Bordeaux sur une période de 2 ans. Ces diagnostics doivent absolument être réalisés précocement pour différentes raisons : – réaliser une surveillance adaptée. La connaissance de l’évolution et des risques de ces pathologies permettent un prise en charge multidisciplinaire afin de prévenir certaines complications (cardiaque, ophtalmologique, orthopédique, etc.) ; – la possibilité de traitement symptomatique. Les bisphophonates sont fréquemment utilisés dans plusieurs de ces pathologies (maladies métaboliques et ostéogenèse imparfaite) avec de bons résultats. D’autres traitements permettent d’améliorer le confort de ces patients ; – l’existence de traitement étiologique. L’arrivée d’enzymothérapie substitutive dans les maladies lysosomales (maladie de Gaucher, de Fabry et de Hurler à ce jour) nécessite des diagnostics précoces afin de prévenir l’apparition de certaines complications. D’autres thérapies (interféron alpha, anti-TNF et IL 1Ra) peuvent être proposées pour le traitement des maladies autoinflammatoires ; – enfin, en raison de la nécessité d’un conseil génétique. Si la pathologie est de transmission autosomique récessive, le patient n’a le plus souvent aucun risque pour sa descendance en dehors d’une union consanguine. Le diagnostic de maladie de transmission dominante ou lié à l’X nécessitera une explication claire des risques et des moyens de prévention.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :