Dermatologie
Publié le 31 aoû 2006Lecture 9 min
Histiocytoses non langerhansiennes de l’enfant
M. Rybojad - Hôpital Saint-Louis, Paris
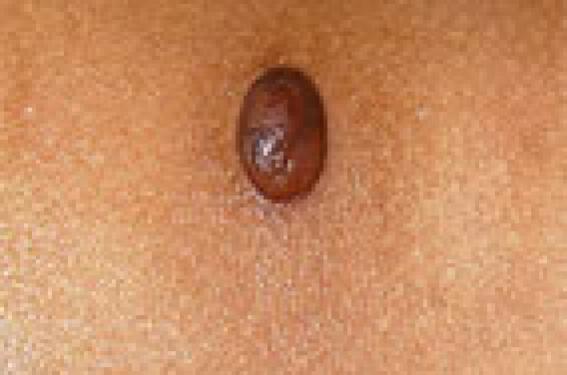
La classification des histiocytoses non langerhansiennes est fondée sur des critères cliniques, histologiques, immunohistochimiques et évolutifs. Toutefois, elle demeure encore sujette à discussion et est compliquée par l’existence de possibles formes de passage entre différentes entités, de cas frontières cliniques et/ou histologiques ou de modification de l’histologie selon le stade évolutif.
Cela, ainsi que la rareté de ces affections, explique que les données thérapeutiques les concernant ne soient pas codifiées. Elles reposent sur des publications de cas isolés ou de petites séries, dont la lecture et l’interprétation peuvent être rendues difficiles par la variabilité des dénominations employées pour désigner les différents types d’histiocytoses non langerhansiennes.
Nous excluerons de ce cadre les proliférations histiocytaires secondaires à une autre pathologie (infectieuse, immunitaire, etc.) ou d’origine métabolique, pour n’aborder que le cadre des histiocytoses non langerhansiennes primitives. Avant d’aborder l’étape thérapeutique, il est important de définir mieux le type de l’histiocytose. Éliminer une histiocytose langerhansienne ne pose, dans la majorité des cas, pas de problème, mais il faut s’efforcer de typer au mieux l’histiocytose non langerhansienne, 1’évolution et le pronostic, étant variable d’une entité à l’autre, ce qui modifie par conséquent l’attitude thérapeutique. Le diagnostic repose sur des données cliniques telles que l’âge de survenue, la distribution et la taille des lésions, l’association à des signes viscéraux, et sur la biopsie cutanée. Celle-ci doit comporter une étude histologique conventionnelle (aspect des histiocytes, topographie de l’infiltrat – dermique dans les histiocytoses non langerhansiennes –, association à d’autres cellules, surcharge lipidique) ainsi qu’une analyse phénotypique par immunohistochimie (protéine S100, antigènes CDla et CD68). Figure 1. Xanthogranulome juvénile de la face postérieure de la jambe chez un enfant de 8 ans. Figure 2. Xanthogranulome juvénile. Le traitement est très variable en raison de l’hétérogénéité des situations en cause. Certaines sont autorégressives et peu affichantes et n’imposeront pas de traitement, d’autres sont théoriquement au contraire régressives mais affichantes. Elles peuvent aussi être localisées et être traitées chirurgicalement. D’autres, enfin, sont non régressives ; en outre, certaines de ces affections sont associées à d’autres pathologies (diabète insipide, etc.) qui imposeront leur traitement propre. Xanthogranulome juvénile C’est la plus fréquente des histiocytoses non langerhansiennes. Elle survient le plus souvent chez le nourrisson et l’enfant (80 % avant 2 ans) et son évolution est le plus souvent purement cutanée. La répartition est la même dans les deux sexes. La physiopathologie n’est pas élucidée. Le xantogranulome juvénile est la plus fréquente des histiocytoses non langerhansiennes. Elle se caractérise par un ou plusieurs papulonodules jaune orangé dont la taille varie de quelques millimètres à 1 ou 2 cm, situés à la partie supérieure du corps et exceptionnellement sur les muqueuses. Formes cliniques La forme micronodulaire est la plus fréquente (60 %) et peut s’associer à des taches café au lait dans le cadre d’une neurofibromatose de type I. Une surveillance dans le cadre d’une leucémie myélomonocytaire doit être effectuée. La forme macronodulaire peut s’associer à une atteinte systémique (poumons, os, reins, testicule, péricarde, etc.). Celle-ci ne s’exprime pas sur le plan clinique et régresse habituellement en quelques années. D’autres formes cliniques ont été rapportées, notamment des xanthogranulomes juvéniles (XGJ) géants ou sous-cutanés à croissance rapide. L’aspect clinique est atypique, multinodulaire, infiltrant et de très grande taille. Ces signes sont à l’origine d’un problème diagnostique imposant d’éliminer les autres tumeurs néonatales : fibromatose infantile ou tumeurs au pronostic plus péjoratif, notamment une tumeur de Darier-Ferrand, un fibrosarcome infantile, un rhabdomyosarcome ou un sarcome embryonnaire. Dans ces cas, la biopsie avec immunomarquage permet de porter le diagnostic et de proposer une abstention thérapeutique avec surveillance régulière. Dans 10 % des cas de XGJ, une atteinte oculaire spécifique est à redouter ; elle touche principalement l’iris avec une néovascularisation responsable de phénomènes hémorragiques (hyphema de la chambre antérieure de l’œil). La gravité potentielle de cette atteinte qui peut être insidieuse et parfois passer inaperçue au début, justifie la réalisation d’un examen ophtalmologique systématique. Il n’y a pas de perturbation du métabolisme lipidique. Histologie Histologiquement, il existe un infiltrat dermique de cellules épithélioïdes avec des histiocytes vacuolisés et spumeux ou cellules de Touton. L’immunomarquage retrouve un infiltrat de cellules S100 -, CD1a -, CD68 +. Au stade tardif, la fibrose prédomine alors que la xanthomatisation disparaît. Evolution La régression spontanée est habituelle en quelques mois voire quelques années, pouvant laisser une cicatrice atrophique ou hyperpigmentée. Une fois le diagnostic confirmé, aucun traitement n’est nécessaire sauf en cas d’atteinte oculaire où une corticothérapie générale ou intraoculaire sous-conjonctivale peut être nécessaire pour éviter une hypertonie de la chambre antérieure de l’œil, une uvéite antérieure, des synéchies postinflammatoires et surtout un glaucome par fermeture de la chambre antérieure de l’œil (risque de cécité définitive). Figure 3 (A gauche). Xanthogranulome juvénile. Cellule de Touton au centre du champ. Figure 4 (A droite) . Histiocytose de Rosai-Dorfman : espace lymphatique contenant des histiocytes lymphophagocytotiques caractéristiques (H & E, x 240). Histiocytose céphalique bénigne Décrite par Gianotti et coll. en 1971, il s’agit d’une affection rare. La plupart des cas rapportés dans la littérature concernent des sujets d’origine méditerranéenne. La physiopathologie n’est pas élucidée, mais pourrait faire intervenir un agent environnemental ou infectieux. Elle réalise une éruption d’éléments papuleux jaunâtres, érythémateux ou couleur peau normale, mesurant 2 à 3 mm de diamètre, touchant l’extrémité céphalique (visage, oreilles, cuir chevelu), le cou, s’étendant parfois aux épaules, aux membres supérieurs et au tronc, respectant les muqueuses. Il n’y a pas d’altération de l’état général ni d’atteinte systémique. L’évolution se fait vers la régression spontanée. Celle-ci débute entre 6 mois et 3 ans. La guérison totale sans cicatrices est obtenue en 4 à 9 ans. L’examen histologique avec immunohistochimie retrouve un infiltrat histiocytaire sans cellules spumeuses ou de Touton, infiltrat S100 - et CD1a -. En microscopie électronique, 20 % des histiocytes contiennent des corps en virgule (inclusions) et pas de granules de Langerhans. Le diagnostic différentiel se pose avec le XGJ dans sa forme micronodulaire (papulonodules jaunâtres, présence de cellules géantes et multinucléées en histologie), l’histiocytose langerhansienne (présence de papules squameuses et croûteuses du cuir chevelu et du tronc, positivité des marqueurs des cellules de Langerhans), l’histiocytome éruptif généralisé (aspect histologique comparable, mais absence de prédominance céphalique, possibilité d’atteinte muqueuse, survenue chez l’adulte). L’évolution est bénigne. Aucun traitement n’est nécessaire. Hystiocytose sinusale de Rosai-Dorfman Cette affection rare touche l’enfant mais surtout l’adulte jeune. Elle associe à des signes généraux et de très volumineuses adénopathies cervicales une atteinte cutanée inconstante. Celle-ci est constituée d’éléments papulonodulaires plus ou moins nombreux siégeant en règle au niveau cervico-céphalique. D’exceptionnelles formes cutanées pures ont été rapportées. La biopsie ganglionnaire montre un infiltrat granulomateux d’histiocytes pâles et spumeux, souvent multinucléés regroupés en amas comme des sinus ganglionnaires. Des images d’érythropoïèse sont fréquentes. Il existe un syndrome inflammatoire, une anémie et une hypergammaglobulinémie polyclonale. La régression spontanée est la règle en quelques mois ou plusieurs années. Aucun traitement n’a montré son efficacité. La corticothérapie générale n’apporte qu’une amélioration transitoire et les adénopathies ne sont pas toujours radiosensibles. La vinblastine et l’étoposide ou le méthotrexate peuvent être efficaces, mais doivent être réservés à des formes très évolutives et retentissent sur l’état général. Xanthomatose disséminée de Montgomery (xanthoma disseminatum) Il s’agit d’une affection rare associant une xanthomatose cutanée prédominant aux plis de flexion, des xanthomes muqueux du tractus respiratoire haut, un diabète insipide sensible à la pitressine et une absence d’hyperlipémie. Les xanthomes cutanés sont caractéristiques par leur caractère symétrique et la localisation au niveau des plis des coudes, des creux axillaires, des plis inguinaux et interfessier, des faces latérales du cou et du pourtour buccal et périorbitaire. Les lésions sont constituées d’un semis de papules de consistance ferme, brunes ou chamois, de 2 à 3 mm de diamètre, qui forment au niveau des grands plis, des plaques saillantes. Les lésions muqueuses se situent surtout au niveau buccal et pharyngolaryngé, plus rarement au niveau trachéobronchique. Le métabolisme lipidique est normal. Le diabète insipide est présent dans 30 % des cas et peut précéder de plusieurs années l’apparition des lésions cutanées. Il est lié à l’infiltration xanthomateuse de la posthypophyse. Le processus xanthomateux peut, en outre, intéresser poumon, cœur, vaisseaux, reins, rate, foie et glandes surrénales. Histologiquement, il existe un granulome inflammatoire polymorphe au sein duquel apparaissent des cellules xanthomateuses à cytoplasme spumeux (cellules de Touton). Dans un second temps s’installe une fibrose. Il n’existe aucune anomalie cytologique. L’immunohistochimie montre le caractère non langerhansien de la prolifération histiocytaire. L’évolution est chronique et le retentissement esthétique et fonctionnel est au premier plan, sauf quand l’atteinte des voies respiratoires supérieures est prépondérante. Le traitement repose sur la vasopressine en cas de diabète insipide. Le traitement des xanthomes est difficile ; la corticothérapie générale pourrait prévenir la récidive des xanthomes après chirurgie. Histiocytose mucineuse progressive héréditaire Décrite en 1988 par Bork et Hoede, cette affection autosomique dominante est une histiocytose non langerhansienne. Les lésions papuleuses rouge-brun siègent sur le nez, les mains, les avant-bras, les cuisses. Il n’y a pas d’atteinte viscérale. Les lésions n’ont pas d’évolution spontanément régressive. Histologiquement, il s’agit d’une prolifération d’histiocytes dendritiques produisant de la mucine, comportant dans leur cytoplasme des corps myélinisés et des vacuoles lycopodiacées. Ces cellules appartiennent à la lignée des monocytes-macrophages.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :





