Publié le 09 nov 2009Lecture 9 min
Foie et maladies métaboliques de l'enfant : des grandes lignes pour comprendre
P. LABRUNE, Centre de référence Maladies héréditaires du métabolisme hépatique, service Pédiatrie, hôpital Antoine-Béclère, Clamart. INSERM U 948.
Le foie est un organe central de l’organisme. Il synthétise un très grand nombre de protéines. Il assure la détoxification de différents produits du métabolisme, la régulation des équilibres hormonaux. Il est capable de répondre à différentes stimulations hormonales, en fonction des situations physiologiques dans lesquelles l’organisme se trouve. De nombreuses maladies métaboliques peuvent entraîner des manifestations hépatiques.
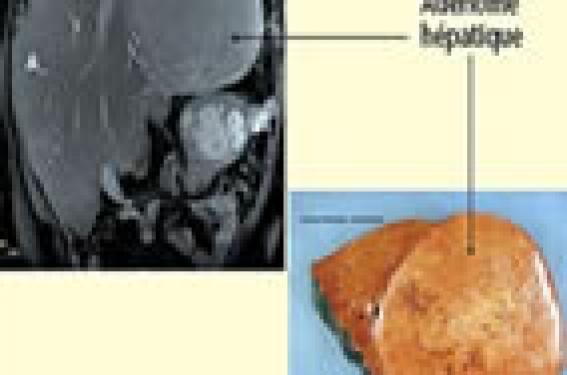
Cytolyse et insuffisance hépato-cellulaire Chez le nouveau-né et le nourrisson, cette association peut correspondre à plusieurs diagnostics : galactosémie congénitale, intolérance héréditaire au fructose, tyrosinémie de type I, hémochromatose périnatale, maladie de la chaîne respiratoire mitochondriale. Chez l’enfant plus grand, on évoque une maladie de Wilson, un déficit en alpha-1-antitrypsine et, plus rarement, une intolérance héréditaire au fructose, voire une galactosémie. La galactosémie congénitale Cette maladie rare se transmet sur un mode autosomique récessif. On estime que, chaque année, environ 11 à 12 enfants naissent en France atteints de cette maladie. La révélation est habituellement néonatale. À la fin de la première semaine de vie, on note un syndrome hémorragique, un ictère, un gros foie, parfois une infection à E. coli. Le diagnostic est urgent et repose sur le Spot test et, surtout à l’heure actuelle, sur la biologie moléculaire qui identifie la ou les mutations au sein du gène codant la galactose-1 phosphate- uridyltransférase. Le traitement est urgent, symptomatique et diététique. Le pronostic à long terme est plus réservé, la survenue d’un retard mental étant fréquente (1). Si l’enfant a dû bénéficier d’une exsanguino-transfusion, les examens moléculaires sont faisables chez ses parents. Le traitement de la galactosémie congénitale est urgent, symptomatique et diététique. L’intolérance héréditaire au fructose Elle se manifeste par des vomissements lors de l’introduction d’aliments saccharosés. Si la prise de ces aliments est répétée, les signes peuvent s’aggraver, associant un ictère, un syndrome hémorragique, une hypoglycémie, un gros foie. Souvent, le diagnostic est plus difficile à faire, car les parents attentifs ont noté que chaque tentative d’ingestion de jus de fruits entraînait des vomissements et ont alors spontanément supprimé le sucre de l’alimentation de leur enfant. Ce dernier ne s’en est pas plaint puisqu’il a, assez régulièrement, un dégoût spontané pour le sucre. Parfois, l’intolérance héréditaire au fructose est une complication iatrogénique. Cela peut être le cas lorsque, le diagnostic n’étant pas connu, l’enfant est hospitalisé pour une affection aiguë et qu’il est réalimenté ou réhydraté avec des produits contenant du fructose. Il faut tout particulièrement se méfier des solutés de réhydratation orale qui contiennent quasiment tous du fructose. Il faut également prendre garde aux médicaments dont les excipients contiennent du fructose. L’intolérance au fructose peut être révélée par une complication iatrogénique. Le diagnostic repose sur l’identification de la ou des mutations en cause au sein du gène codant la fructose-1- phosphate aldolase (2). Il n’y a pas d’indication à un diagnostic prénatal, mais il est tout à fait faisable et souhaitable de faire le diagnostic pré-symptomatique chez les membres de la fratrie afin de juger de l’opportunité ou non de les mettre au régime. Ce régime n’est habituellement pas contraignant, les enfants ayant en général un dégoût spontané pour le fructose. Une supplémentation en vitamine C doit être apportée. L’hémochromatose périnatale C’est une affection rare, liée à une allo-immunisation maternofoetale. Elle se manifeste par une insuffisance hépato-cellulaire néonatale extrêmement sévère, avec des perturbations du bilan martial (hyperferritinémie). Lorsque l’imagerie peut être faite, elle met en évidence une surcharge martiale dans de nombreux organes, dont le foie et les glandes salivaires. Le pronostic est le plus souvent très sombre. Le risque de récidive est majeur, mais des études récentes ont mis en évidence l’efficacité du traitement préventif par perfusion d’immunoglobulines chez la mère à partir de la 18e semaine d’aménorrhée (3). Le pronostic de l’hémochromatose périnatale est le plus souvent très sombre. Cholestase Une cholestase peut être révélatrice de nombreuses affections : déficit en alpha-1-antitrypsine, mucoviscidose, cholestase intrahépatique familiale progressive, maladie de la chaîne respiratoire mitochondriale, maladie péroxysomale, maladie de Niemann- Pick, maladie de la synthèse des acides biliaires, maladie touchant la glycosylation des protéines (CDG syndrome). Cliniquement, une hépatomégalie peut être le signe d’appel. Il faut alors préciser si l’hépatomégalie est isolée ou associée (cholestase, cytolyse). La discussion est différente en fonction de l’âge de l’enfant, des caractères de l’hépatomégalie, de l’association ou non à une splénomégalie (4). Lorsque l’hépatomégalie s’associe à une hyperbilirubinémie conjuguée, certaines maladies métaboliques doivent être évoquées. Une cholestase intra-hépatique doit faire rechercher, outre une galactosémie précédemment étudiée, un déficit en alpha-1-antitrypsine, une tyrosinémie, une maladie de surcharge (5). Il faut néanmoins rappeler que la première cause de cholestase néonatale est l’atrésie des voies biliaires extra-hépatiques qui touche 1 nouveau-né sur 10 000. Le diagnostic est urgent (hépatomégalie, selles décolorées, urines foncées). Le traitement chirurgical doit être effectué avant la 6e semaine de vie pour avoir le maximum de chances d’efficacité. La première cause de cholestase néonatale est l’atrésie des voies biliaires extra-hépatiques. Le déficit en alpha-1-antitrypsine Il peut être à l’origine d’une cholestase néonatale prolongée. Le diagnostic repose sur l’électrophorèse des protides qui montre l’absence de pic d’alpha-1-globuline, sur le dosage pondéral de l’alpha-1-antitrypsine et l’étude du phénotype PI. Ces enfants doivent être surveillés car certains peuvent voir leur atteinte hépatique évoluer vers la cirrhose. Les maladies de surcharge Ces affections associent une hépatomégalie, une splénomégalie et parfois un ictère. Cliniquement, on peut également trouver des signes d’atteintes pulmonaires, osseuses, cérébrales. C’est par exemple le cas de la maladie de Niemann-Pick de type B, liée à un déficit en sphingomyélinase. Outre l’hépatosplénomégalie, cette maladie s’accompagne d’une atteinte pulmonaire interstitielle. L’histologie hépatique met en évidence des cellules de surcharge. Ces dernières sont également retrouvées lors de l’étude de la moelle osseuse. Les mucopolysaccharidoses Adénome hépatique au cours de l’évolution d’une glycogénose de type I. A : aspect en IRM ; B : aspect de la pièce opératoire (cliché du Dr A.C. Mas, Service d’anatomie pathologique, hôpital Antoine-Béclère). Elles peuvent également être à l’origine d’une hépatosplénomégalie. C’est le cas de la maladie de Hunter qui, chez un garçon, associe, outre l’hépatosplénomégalie, une dysmorphie faciale, un retard des acquisitions, une myocardiopathie, une atteinte ostéoarticulaire. L’expressivité de cette maladie est variable. De la même manière, la maladie de Hurler, ou Hurler-Scheie, associe à une hépatosplénomégalie une dysmorphie faciale, une atteinte osseuse, cardiaque et cornéenne. La variabilité d’expression est grande. Un traitement substitutif par enzyme recombinante est disponible pour cette maladie. Chez un grand enfant ou un adolescent, la constatation d’une hépatomégalie hétérogène sur l’examen tomodensitométrique, associée à une hypertransaminasémie prolongée, à une hémolyse compensée, à une tubulopathie, doit faire évoquer le diagnostic de maladie de Wilson. Il faut alors faire rechercher un anneau cornéen de Kayser-Fleischer par l’examen ophtalmologique à la lampe à fente, évaluer la cuprurie, doser la céruloplasmine. L’étude moléculaire peut être intéressante. Le traitement est urgent. L’existence d’une hépatomégalie molle, associée assez souvent à l’impression de « grosses joues bien rebondies », doit faire évoquer une glycogénose, ce d’autant que surviennent des hypoglycémies de jeûne court. Si lors de cette hypoglycémie, on constate une acidose lactique, il s’agit plutôt d’une glycogénose de type I (déficit en glucose-6- phosphatase). S’il n’y a pas d’acidose lactique, il s’agit plutôt d’une glycogénose de type III (déficit en amylo-1-6-glucosidase). Une hépatomégalie molle, associée à l’impression de grosses joues et à des hypoglycémies de jeûne, doit faire évoquer une glycogénose. Chez un enfant atteint de glycogénose de type I, le foie grossit durant les premiers mois. Il peut encore grossir après, surtout si l’équilibre métabolique n’est pas satisfaisant. Il reste mou et peut être le siège du développement d’adénomes multiples, au-delà de la 10e voire de la 15e année de vie(6) (figure). Chez un enfant atteint de glycogénose de type III, le foie peut parfois devenir fibreux à mesure que l’âge avance. Les adénomes surviennent de façon beaucoup plus exceptionnelle. Le risque de cirrhose est faible mais non nul. À cette atteinte hépatique s’associe le plus souvent une atteinte musculaire (6). Conclusion Il faut rappeler l’importance du contexte clinique (âge, association ou non à une splénomégalie, à une cholestase). La présence de signes associés (os, poumon, coeur, cerveau) est également un élément qu’il faut prendre en compte. Les examens d’imagerie (échographie, scanner, IRM) ont une grande importance et ne doivent être effectués, tout comme les examens biologiques, qu’en fonction de l’orientation clinique.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :