Publié le 28 juil 2008Lecture 8 min
Comment diagnostiquer en pratique une photodermatose congénitale ?
H. ADAMSKI, Service de Dermatologie, CHU Pontchaillou, Rennes
Les photodermatoses congénitales sont des dermatoses héréditaires se manifestant par une photosensibilité. Elles peuvent être soit liées à une déficience de la protection cutanée naturelle aux ultraviolets, soit d’origine métabolique, responsables de la présence anormale de molécules photosensibilisantes.
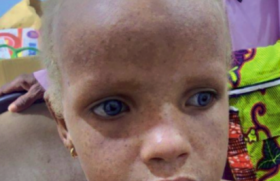
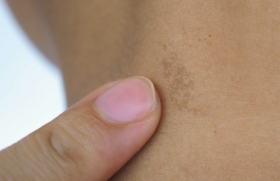
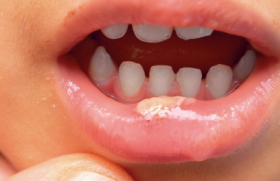
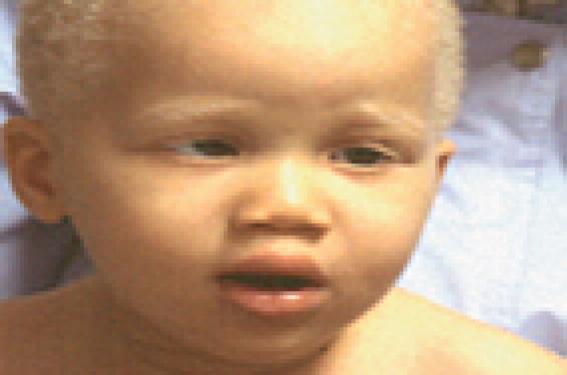
Figure 1. Albinisme oculo-cutané. Manifestations cliniques cutanées Photosensibilité La photosensibilité se manifeste par une réaction cutanée anormale sur les zones habituellement découvertes (visage, cou, décolleté, mains) après une exposition solaire. Dans le cadre des photodermatoses congénitales, elle se traduit par un phénomène de phototoxicité qui est récidivant après une exposition, même minime. L’aspect cutané est celui d’un « coup de soleil » ou érythème actinique, s’accompagnant de sensation de brûlure pouvant être suivi d’oedème et parfois de bulles. L’évolution peut se faire vers une pigmentation durable. Leucodermie Elle correspond à une décoloration de la peau diffuse ou localisée résultant de la disparition soit totale, soit partielle de la mélanine, réalisant alors soit une achromie, soit une hypochromie. Dans les formes sévères de leucodermie, la peau devient sensible aux ultraviolets par inaptitude au bronzage. Génophotodermatoses avec leucodermie Figure 2. Vitiligo. Elles sont dues à des anomalies de la formation ou de la distribution de la mélanine (1). Albinismes oculo-cutanés (2) Ils réalisent des hypochromies généralisées cutanées, pilaires et oculaires. Le diagnostic est clinique et souvent évident à la naissance (figure 1). Différents types cliniques ont été identifiés, tous de transmission autosomique récessive. Dans les formes sévères, il existe une très forte sensibilité cutanée au soleil par une inaptitude au bronzage et l’apparition précoce d’une héliodermie et de carcinomes cutanés. Au niveau cutané, le nombre de mélanocytes est normal. Plusieurs gènes sont impliqués dans la pathogénie des albinismes oculo-cutanés codant soit des protéines intervenant dans la mélanogenèse (tyrosinase, TRP1), soit des protéines à fonction mal définie (MATP, par ex.). Dans de rares cas, les albinismes peuvent être associés à d’autres symptômes : syndrome de Chediak-Higashi (présentant également un déficit fonctionnel du polynucléaire), syndrome de Hermansky-Pudlak (qui associe des troubles de coagulation) et syndrome de Griscelli- Pruniéras (thrombopénie et anomalie neurologique). Le diagnostic différentiel des albinismes est représenté essentiellement par les dilutions pigmentaires regroupant les affections métaboliques héréditaires où la leucodermie est discrète. La symptomatologie cutanée n’est qu’un élément contingent à d’autres manifestations, en particulier neurologiques : maladie de Menkes, phénylcétonurie... Vitiligo C’est une affection fréquente (environ 1 %) due à une disparition des mélanocytes. Elle n’est déterminée génétiquement que dans un tiers des cas. Le vitiligo se présente généralement par des plages hypopigmentées bien limitées, soulignées par une bordure hyperpigmentée. Il peut apparaître à tout âge, mais les premiers signes surviennent le plus souvent chez l’adulte jeune (figure 2). Figure 3. Xeroderma pigmentosum. Piébaldisme Il est caractérisé par une mèche blanche, prolongée d’une achromie frontale, souvent associée à des macules dépigmentées thoraciques et sur les membres en épargnant les extrémités. Le piébaldisme est un des éléments du syndrome de Waardenburg, qui se caractérise également par une dysmorphie faciale, surdité de perception et hétérochromie irienne. Génophotodermatoses sans leucodermie Photodermatoses avec déficience de la réparation de l’ADN(3) Xeroderma pigmentosum (4) Il s’agit d’une affection rare caractérisée par une photoxicité survenant dès les premiers mois de vie, suivi d’un état poïkilodermique (atrophie cutanée avec achromie ou pigmentation et télangiectasies) des zones découvertes (figure 3). Ces lésions dégénèrent en tumeurs cutanées (carcinomes et mélanomes) vers l’âge de 4 ans, avec une fréquence 4 000 fois plus élevée que dans la population normale. D’autres manifestations cliniques sont associées, comme des neuropathies et des anomalies ophtalmologiques (photophobie, kératite…). C’est une maladie héréditaire transmise sur le mode autosomique récessif et liée à un déficit des systèmes enzymatiques de réparation des lésions de l’ADN par les ultraviolets. Huit gènes ont été identifiés pour la forme classique. Il existe une variante du xeroderma pigmentosum qui se révèle par une photosensibilité moins marquée et la survenue plus tardive des néoplasies cutanées, où à ce déficit de réparation de l’ADN, il s’associe un déficit en catalase (enzyme antioxydante). Dans ce cadre s’ajoute d’autres maladies de transmission autosomique récessive et exceptionnelles par leurs fréquences : Figure 4. Syndrome de Cockayne. PIBIDS syndrome Ce syndrome est un type de trichothiodystrophie caractérisée par une photosensibilité (Photosensitivity), une ichtyose (Ichtyosis), des cheveux cassants (Brittle hair), un déficit intellectuel (Intellectual impairement), une hypofertilité (Decrease fertility) et un nanisme (Short stature). Au niveau génétique, il est retrouvé le même défaut de réparation de l’ADN que dans le xeroderma pigmentosum. Mais le PIBIDS syndrome ne s’accompagne pas de cancers cutanés pouvant être expliqués par l’absence de déficit en catalase associé retrouvée(5). Syndrome de Cockayne L’affection débute vers 6 mois par une photosensibilité. Il s’y associe une microcéphalie e t de grandes oreilles aboutissant au « faciès de Mickey » caractéristique à 2 ans (figure 4). Un nanisme et une encéphalopathie sont également retrouvés. L’évolution est fatale à brève échéance. Syndrome de Bloom Il se manifeste par une photosensibilité évoluant vers une poïkilodermie donnant un aspect pseudo-lupique du visage et un retard staturo-pondéral. Des taches café au lait sont retrouvées dans la moitié des cas. Il existe un déficit immunitaire variable prédisposant aux hémopathies malignes. Syndrome de Rothmund-Thomson Les manifestations cliniques surviennent vers 3 mois par un érythème télangiectasique, avec atrophie du visage puis des membres et des fesses. La photosensibilité est habituelle et s’atténue avec l’âge. Le faciès est particulier, avec un front haut et un nez effilé (figure 5). La cataracte est fréquente. Cette affection s’associe à une incidence plus élevée de carcinomes cutanés et d’ostéosarcome. Figure 5. Syndrome de Rothmund-Thomson. Photodermatoses métaboliques (6) Figure 6. Protoporphyrie érythropoïétique. Porphyries (7) Les porphyries regroupent un ensemble de maladies caractérisées par un déficit sur la voie de la synthèse de l’hème. La conséquence en est l’accumulation tissulaire de substances photosensibilisantes que sont les porphyrines. La plupart des porphyries sont congénitales, souvent de transmission autosomique dominante, avec une pénétrance variable. Elles peuvent être classées suivant leurs manifestations cliniques. Parmi les porphyries cutanées à révélation infantile, on insistera sur la protoporphyrie érythropoïétique et la maladie de Günther. • La protoporphyrie érythropoïétique est la porphyrie la plus fréquente de l’enfant. Elle devrait être recherchée en cas des « brûlures cutanées » souvent associées à des plaques oedémateuses survenant lors des expositions solaires (figure 6). L’évolution est habituellement favorable, avec régression des poussées à l’âge adulte. Cependant, l’atteinte hépatique est présente dans 25 % des cas environ et conditionne le pronostic. La principale complication est la cholestase. Dans 1 % des cas environ, l’évolution se fait vers la constitution d’une cirrhose(8). • La maladie de Günther ou porphyrie érythropoïétique congénitale est la porphyrie cutanée la plus grave et s’exprime dès les premiers mois de vie. La photosensibilité est importante, aboutissant à des cicatrices rétractiles et à des mutilations des extrémités. Maladie de Hartnup Il s’agit d’une maladie très rare, à transmission autosomique récessive, liée à un trouble d’absorption digestive et de réabsorption rénale des acides aminés neutres, dont le tryptophane. Le tableau associe un érythème violacé, avec desquamation des zones découvertes. Au bout de plusieurs poussées, la peau est pigmentée. Il s’y associe des troubles neurologiques pouvant être sévères (ataxie cérébelleuse, confusion). Le diagnostic repose sur la chromatographie des acides aminés urinaires, mettant en évidence une aminoacidurie et une élévation du tryptophane. Conclusion Le diagnostic précoce des photodermatoses congénitales est fondamental afin d’optimiser leurs prises en charge. Un suivi régulier sera alors réalisé pour vérifier la bonne observance des mesures de photoprotection et aussi pour le dépistage des cancers cutanés et des complications viscérales.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :