Publié le 25 nov 2024Lecture 5 min
Une induration qui dure
Mustapha MAZEGHRANE, Centre hospitalier André Grégoire, Montreuil
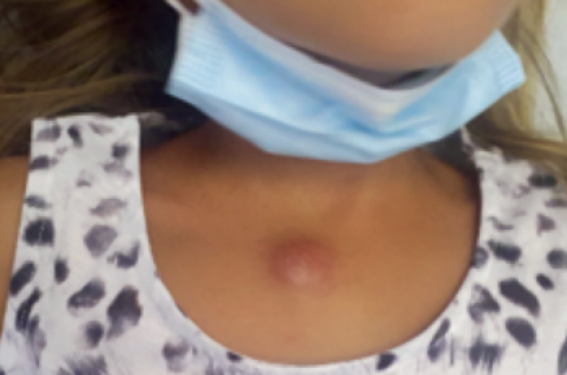
Il s’agit d’une jeune fille de 7 ans, premier enfant d’un couple non apparenté sans antécédents personnels ou familiaux pathologiques, qui a présenté une tuméfaction sternale de 3 cm d’apparition progressive. L’état général de l’enfant est conservé, et elle n’a jamais été fébrile ni algique. Un voyage à Madagascar a été rapporté par la famille deux mois avant l’apparition de la lésion.
Devant cet aspect de pseudo-abcès, un traitement par antibiothérapie orale de 10 jours est débuté en ville, sans complément d’examen. Malgré une bonne compliance, la lésion ne cesse de grossir. Après un mois, elle est devenue peu à peu douloureuse et les parents décident de consulter aux urgences.
Consultation aux urgences
Aux urgences, la jeune fille est apyrétique, peu algique, avec un état général conservé. Un bilan biologique est alors réalisé : NFS avec 13 000 GB/mm³, dont 4 100 PN/mm³ et 470/mm³, une CRP à 10 mg/l ; augmentation modérée des transaminases, les phosphatases alcalines sont normales (145 UI/l) ; LDH 220 UI/l.
Une échographie objective une lésion tissulaire nodulaire, hypervascularisée d’échostructure hypoéchogène hétérogène, en forme de sablier, centrée de part et d’autre du manubrium sternal.
Un scanner est réalisé et révèle une lyse osseuse sternale. Une IRM montre une lésion prenant le contras te avec infiltration des tissus mous.
Les hypothèses à ce stade s’orientent vers une ostéomyélite chronique infectieuse (staphylocoque, tuberculose osseuse, infection à Kingella kingae) ou une lésion tumorale (sarcome, métastase d’un neuroblastome, ostéome).
Une biopsie est effectuée et met en évidence un liquide comportant des fausses membranes purulentes. Une antibiothérapie IV (intraveineuse) est initiée pendant plusieurs semaines mais interrompue car toutes les cultures sont revenues négatives. Les PCR et cultures BK sont également négatives, de même que la PCR Kingella kingae.
L’examen anatomopathologique révèle quant à lui une lésion d’histiocytose langerhansienne avec nécrose, sans arguments en faveur d’un sarcome.
Le bilan d’extension incluant PETscan et scintigraphie osseuse n’apporte pas plus d’information dans le diagnostic retenu, celui d’une histiocytose langerhansienne unifocale osseuse.
Un traitement par AINS est alors débuté et maintenu pendant 20 jours, et une régression complète est observée en quelques semaines.
Discussion
L’histiocytose est définie par une prolifération des cellules du système mononucléaire, appelées cellules de Langerhans lorsqu’elles se trouvent dans l’épiderme ou histiocytes dans le tissu conjonctif. Les dysfonctionnements des histiocytoses sont multiples et variés. Chez l’enfant, l’incidence est rare et est estimée à 1 cas sur 200 000 par an(2). Le nombre de nouveaux cas est estimé à environ 50 par an chez les enfants en France. La durée de la maladie est difficile à prévoir puisqu’elle varie de quelques semaines pour certaines formes (atteinte osseuse unique) à plusieurs mois. Il existe des formes à rechute. De nombreuses classifications ont été proposées selon la localisation, l’extension, les critères histologiques incluant l’immunophénotypage. Selon l’Histiocyte Society(1), on distingue en effet trois groupes d’histiocytoses :
– classe 1 : l’histiocytose de Langerhans unifocale ou multisystémique ;
– classe 2 : les histiocytoses non langerhansiennes bénignes (xanthogranulome juvénile, maladie de Rosai-Dorfmann, maladie de Erdheim-Chester, lymphohistiocytose hémophagocytaire) ;
– classe 3 : les maladies histiocytaires malignes (histiocytose maligne, sarcome histiocytaire) qui touchent essentiellement l’adulte.
Présentation clinique
Cette maladie est hétérogène dans sa présentation clinique. Le diagnostic peut être posé à tout âge, de la naissance à l’adolescence, avec un pic de fréquence entre 1 et 3 ans(1), et un sex-ratio de 1/2. L’étiopathogénie de l’histiocytose X reste inconnue.
La localisation peut être ubiquitaire. Les anomalies osseuses sont les plus fréquentes, 60 % des cas rapportés ne touchent que l’os, notamment le crâne, la face et les côtes(3,4). L’imagerie des structures osseuses touchées peut évoquer le diagnostic devant une ostéolyse bien limitée, entourée d’une ostéosclérose périphérique, unique ou multiple. Il est beaucoup plus difficile de diagnostiquer une localisation unique ou rare nécessitant le recours à l’étude histologique.
Les atteintes dermatologiques sont constatées dans 30 % des cas. Il s’agit de nodules ou d’érythèmes vésiculopustuleux étendus sur le tronc (pseudo-varicelle), la région axillaire et inguinale, parfois des croûtes de lait sur le cuir chevelu des nourrissons. Au niveau du poumon, elle peut se manifester par une gêne respiratoire et, en radiographie, par un infiltrat ou des bulles. Si elle atteint le tube digestif, elle peut entraîner une hépatosplénomégalie souvent de mauvais pronostic.
Les localisations endocriniennes sont exceptionnelles, mais peuvent entraîner un diabète insipide. Les localisations neurologiques sont également exceptionnelles, elles se révèlent par des signes pseudotumoraux ou des signes neurodégénératifs, tels que des troubles cognitifs, de l’attention, des troubles moteurs ou un syndrome cérébelleux.
La maladie peut ou non se compléter dans le temps.
Prise en charge
Concernant la prise en charge, le traitement doit être adapté à la sévérité de la maladie et à son profil évolutif, allant de l’abstention thérapeutique à la chimiothérapie en passant par les traitements anti-inflammatoires locaux ou généraux.
La prise en charge des formes unifocales est le plus souvent limitée avec une évolution bénigne comme chez notre jeune fille. Les localisations osseuses uniques ou peu nombreuses ne nécessitent généralement pas de traitement en dehors de la biopsie pour confirmer le diagnostic et d’un curetage éventuel ou encore d’une injection locale de corticoïdes(5,6).
Évolution
Dans 40 % des cas, les formes localisées guérissent spontanément en quelques semaines à quelques mois. Cependant, il s’agit d’une maladie capricieuse et 30 % des enfants connaissent une récidive locale ou systémique pouvant menacer le pronostic vital ou fonctionnel.
Conclusion
L’histiocytose langerhansienne, bien que répertoriée depuis longtemps sous des noms variés, est une entité dont l’étiopathogénie demeure mystérieuse. Il s’agit d’une maladie principalement pédiatrique, heureusement le plus souvent bénigne. Notre observation souligne l’importance de connaître cette entité. Elle peut prendre divers aspects que le praticien non spécialiste doit reconnaître pour éviter tout retard diagnostique. Ces patients doivent être orientés vers un centre de référence où un diagnostic histologique, un bilan d’extension et un projet thérapeutique adapté pourront être envisagés.
Figure 1. Lésion cutanée à type de nodule évoluant depuis plusieurs semaines.
Figure 2. Échographie montrant un envahissement du sternum.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :