Publié le 31 oct 2023Lecture 6 min
Diarrhées et vomissements chroniques…
Sarah BAMBERGER, Service de pédiatrie, hôpital Delafontaine, Saint-Denis
La rubrique « En direct des staffs » est ouverte à tout médecin d’un service de pédiatrie souhaitant partager avec les lecteurs de Pédiatrie Pratique les cas discutés dans son service et qu’il estime suffisamment intéressants et édifiants pour être portés à la connaissance de ses confrères.
Il s’agit d’un nourrisson de 4 mois consultant aux urgences pour diarrhées et vomissements chroniques. Ce nourrisson est né à 39 SA avec un poids de naissance de 3 300 g. Il s’agit du 4e enfant de parents consanguins, originaires d’Algérie. Les parents rapportent que, dès la 1re semaine de vie, leur fils a présenté une diarrhée avec 2 à 3 selles liquides par jour, de nombreux vomissements, un ballonnement abdominal et un inconfort global. Devant ce tableau, un RGO a été évoqué, amenant à un épaississement des biberons et un traitement par ésoméprazole, sans amélioration. Secondairement, une allergie aux protéines du lait de vache non IgE-médiée a été évoquée et un mélange d’acides aminés a été prescrit. Ce changement de lait a permis de diminuer le nombre de vomissements mais la diarrhée a persisté.
La famille consulte finalement aux urgences à l’âge de 4 mois devant une majoration des symptômes digestifs et une cassure pondérale depuis l’âge de 2 mois. L’examen clinique met en évidence un abdomen distendu, ainsi que des stigmates de dénutrition (masse musculaire faible, PB/PC – périmètre brachial rapporté au périmètre crânien à 0,25). Le bilan aux urgences montre une NFS normale, une acanthocytose sur le frottis, un ionogramme sanguin normal, une albumine à 30 g/l et une cytolyse à 2N sans cholestase. Devant ce tableau digestif chronique avec cassure pondérale, le bilan étiologique est le suivant :
– un bilan infectieux négatif (coproculture, parasitologie des selles, sérologies CMV, EBV, VIH ;
– un bilan métabolique normal (ammoniémie normale, chromatographie des acides aminés plasmatique normale, chromatographie des acides organique urinaire normale) ;
– un bilan neurologique normal avec une IRM cérébrale sans particularité ;
– un bilan thyroïdien avec TSH normale.
Le bilan à orientation digestive a mis en évidence une stéatorrhée à 3 g/24 h, avec une clairance de l’alpha 1 antitrypsine augmentée à 30 ml/24 h, une carence profonde en vitamines D et E.
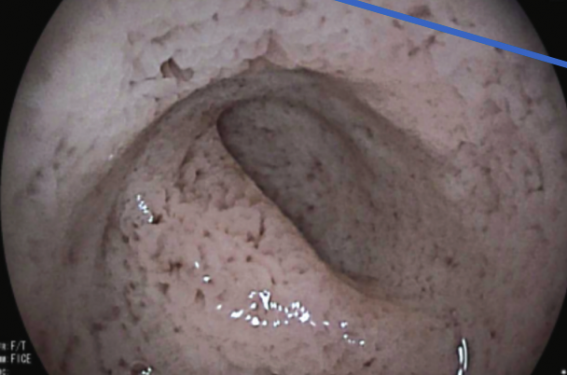
L’enfant est adressé pour une endoscopie œsogastroduodénale. L’examen montre un aspect œdémateux et blanchâtre/laiteux du 2e duodénum (figure 1). L’analyse histologique met en évidence une surcharge entérocytaire au niveau de l’épithélium de surface du D2, sans anomalie du relief villositaire (figure 2). Le frottis sanguin confirme la présence d’une acanthocytose (figure 3). Devant cette surcharge entérocytaire, un bilan lipidique est finalement réalisé et retrouve d’importantes anomalies avec un cholestérol total abaissé à 1,06 mmol/l, des triglycérides indosables, un LDL-cholestérol indosable, ainsi qu’une apolipoprotéine B.
Figure 1. Aspect macroscopiquement anormal du 2e duodénum. Aspect œdémateux/laiteux/blanchâtre.
Figure 2. Biopsie du 2e duodénum. Aspect histologique de surcharge entérocytaire. L’architecture duodénale est normale sans atrophie villositaire. Il existe par contre une surcharge lipidique entérocytaire sous forme de vésicules, prédominant à la partie supérieure des villosités.
Figure 3. Acanthocytose sur le frottis (aspect spiculé des globules rouges).
En l’absence d’argument pour une hypocholestérolémie secondaire (cf. discussion), le diagnostic le plus probable évoqué est celui d’une hypocholestérolémie familiale intestinale. La confirmation génétique est en cours.
Discussion
Les hypocholestérolémies familiales sont des maladies génétiques rares du métabolisme des lipoprotéines. Les lipoprotéines sont des complexes micellaires associant des lipides et des protéines. Elles sont impliquées dans le transport des lipides en phase aqueuse et des vitamines liposolubles A, D, E et K. Elles sont produites par les entérocytes (formant les chylomicrons) et par les hépatocytes (formant les Very Low Density Lipoprotein [VLDL]). L’assemblage des lipoprotéines n’est possible qu’en présence de trois protéines principales : Apoliprotéine B (ApoB), la Microsomal triglyceride transfer protein (MTTP), et la protéine de transport Sar1GTPase. Si l’une de ces trois étapes dysfonctionne il en résulte, au niveau intestinal, une malabsorption lipidique par défaut de fabrication ou de sécrétion de chylomicrons et, au niveau hépatique, une stéatose et un défaut de production des VLDL (figure 4).
Figure 4. Métabolisme intra-érythrocytaire des lipides, d’après le PNDS « Hypocholestérolémie génétique intestinale ».
Dans les hypocholestérolémies génétiques intestinales, il y a un déficit en lipoprotéines riches en ApoB, c’est-à-dire les chylomicrons, VLDL et LDL. Il existe trois entités amenant à une baisse des lipoprotéines riches en ApoB : l’abêtalipoprotéinémie par mutation de MTTP, l’hypobêtalipoprotéinémie par mutation d’ApoB et la maladie de rétention des chylomicrons par mutation de SAR1B.
Le tableau clinique apparaît dès les premières semaines de vie. Le défaut de sécrétion des chylomicrons entraîne une surcharge entérocytaire et une malabsorption lipidique, responsable d’une diarrhée chronique avec stéatorrhée. Il s’y associe des douleurs abdominales, des vomissements et des ballonnements. L’intensité du tableau dépend de la quantité de graisses ingérées. Il apparaît rapidement un retard de croissance et des stigmates cliniques de dénutrition.
La surcharge lipidique hépatique peut entraîner une stéatose hépatique, voire une cirrhose.
Il existe également des signes neurologiques liés à la carence en vitamine E. Cette carence induit des lésions de démyélinisation axonale périphérique. On retrouve une baisse des réflexes ostéotendineux, une faiblesse musculaire, une dysarthrie et une ataxie. La carence en vitamine A peut être à l’origine de complications ophtalmologiques (perte de la vision nocturne, perte de la vision des couleurs, rétinite pigmentaire).
Biologiquement, le bilan lipidique retrouve un cholestérol total bas, en particulier le LDL-cholestérol ; des triglycérides bas qui n’augmentent pas après le repas, une Apo B effondrée. Il y a une stéatorrhée importante. Il faut doser les vitamines A, E et D, et faire un TP pour la vitamine K. On retrouve souvent une carence profonde en vitamine E. Le frottis sanguin décrit une acanthocytose, c’est-à-dire des érythrocytes spiculés en forme d’oursin, témoins de la carence en vitamine E. Enfin, le bilan hépatique peut mettre en évidence une cytolyse sans cholestase. Le diagnostic de certitude est génétique.
La prise en charge consiste à limiter la malabsorption en réduisant l’apport en graisses à moins de 30 % de l’apport énergétique global. La restriction lipidique concerne surtout les acides gras (AG) à chaîne longue qui sont les principaux constituants des chylomicrons. L’apport lipidique sera surtout constitué d’AG à chaîne courte. Ce régime nécessite une supplémentation en acides gras essentiels (AGE) et en vitamines liposolubles, afin de limiter les complications neuro-ophtalmologiques.
Chez un nourrisson présentant une diarrhée chronique avec retard de croissance, les diagnostics souvent évoqués (selon l’âge) sont : une APLV non IgE-médiée, une entéropathie post-infectieuse, une maladie cœliaque ou une cause extradigestive type dysthyroïdie. Si ces diagnostics sont infirmés par les explorations ou par l’échec d’un traitement d’épreuve, il est important de demander un bilan lipidique à jeun.
Lorsque le bilan lipidique retrouve une hypocholestérolémie, il faudra penser à éliminer les causes secondaires d’hypocholestérolémie : APLV, maladie cœliaque, insuffisance pancréatique exocrine, diarrhée infectieuse, insuffisance hépatique, hyperthyroïdie et dénutrition.
Si l’hypocholestérolémie semble primitive, en présence de ces signes digestifs, il faudra : explorer la malabsorption en dosant la stéatorrhée et les vitamines liposolubles ; évaluer le retentissement hépatique, ophtalmologique, hématologique ; et effectuer l’analyse génétique permettant de confirmer le diagnostic d’hypocholestérolémie familiale.
Conclusion
Les hypocholestérolémies intestinales primitives sont des maladies génétiques du métabolisme des lipoprotéines provoquant une malabsorption lipidique, une surcharge entérocytaire et hépatocytaire, une carence en vitamines liposolubles. Ces maladies sont très rares. Elles soulignent cependant l’importance de demander un bilan lipidique chez tout nourrisson présentant une diarrhée chronique avec retard de croissance, pour lequel les diagnostics évoqués par argument de fréquence (APLV, entéropathie post-infectieuse, maladie cœliaque) semblent infirmés.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :