L'œil du médecin d'adolescents
Publié le 05 déc 2022Lecture 8 min
Ayoub, 14 ans, présente une fatigue générale et une perte de poids
Renaud DE TOURNEMIRE, CHU Ambroise Paré, Boulogne-Billancourt
Cette rubrique consacrée à la médecine de l’adolescent est l’occasion de présenter une pratique intégrative et systémique centrée sur l’adolescent mais explorant en même temps les interactions avec la famille, les pairs, les soignants. Le corps de l’adolescent y prend une place importante. Cette rubrique a vocation à accueillir les observations d’internes, chefs de clinique, assistants, pédiatres ou médecins d’adolescents qui le souhaitent, en collaboration avec le responsable (renaud.detournemire@aphp.fr). Aujourd’hui, une observation clinique commentée.
Ayoub, âgé de 14 ans, est admis en unité de médecine de l’ adolescent pour altération de l’état général, avec une fatigue persistante, une perte d’appétit et des difficultés pour s’alimenter.
Histoire de la maladie
Il présente une rougeur oculaire et se plaint de photophobie, de douleurs au niveau de la langue associée à une sensation de sécheresse oculaire et buccale. Il ne présente pas de fièvre, ni de céphalées. On ne retrouve pas de plainte digestive, cutanée ou articulaire associée.
Il aurait perdu 2,5 kg en 2 mois.
Il n’y a pas d’argument dans son histoire personnelle faisant évoquer une infection chronique ou un déficit immunitaire (pas de voyage récent ni d’infection récurrente).
Ses deux parents sont d’origine algérienne et ses grands-parents maternels sont cousins germains.
Sur le plan des antécédents familiaux, sa grand-mère maternelle est suivie pour une maladie de Crohn. Il n’y a pas de notion d’autres maladies auto-immune ou auto-inflammatoire dans la famille, ni de maladie rhumatologique inflammatoire ou de psoriasis.
L’examen clinique initial d’Ayoub met en évidence une hypertrophie des glandes parotidiennes avec douleur à la palpation, des adénopathies cervicales sensibles infracentimétriques, une hypertrophie gingivale, ainsi qu’un torticolis et un trismus avec une ouverture buccale limitée à 3 cm. L’auscultation cardiopulmonaire est sans particularités, de même que l’examen neurologique, articulaire et cutané.
Le bilan biologique initial met en évidence une hémoglobine à 14 g/dl, une hyperleucocytose à 16,7 G/L, des polynucléaires à 13 G/L, des plaquettes à 215 G/L, ainsi qu’une CRP à 177 mg/L.
L’ionogramme sanguin, le bilan rénal et hépatique sont normaux.
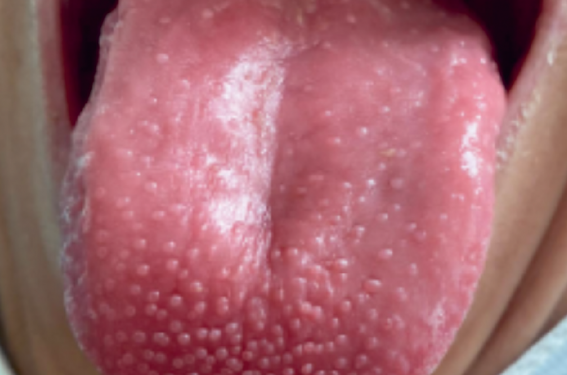
Figure 1. Aspect de langue « rotie » visible dans les syndromes secs.
Évolution
• Sur le plan ORL
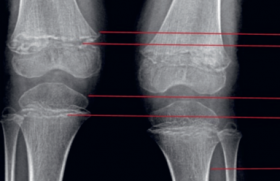
Le scanner cervical (J0, figure 2) met en évidence un aspect hétérogène des glandes sous-maxillaires et parotidiennes, avec quelques dilatations canalaires. L’imagerie ne met pas en évidence de collection profonde, excluant l’hypothèse d’un abcès infectieux.
Devant cette inflammation clinique et biologique, l’hypothèse infectieuse est évoquée, dans un premier temps, et une antibiothérapie probabiliste par voie intraveineuse est débutée par amoxicilline/acide clavulanique à 150 mg/kg/j. Devant l’absence d’amélioration clinique et la majoration du syndrome inflammatoire (CRP à 247 mg/L) à H48, modification de l’antibiothérapie intraveineuse par l’association de ceftriaxone 2 g/j et métronidazole 500 mg toutes les 8 heures devant un doute clinique d’une complication par cellulite.
Le scanner cervical (J2) est contrôlé et l’examen a montré un aspect stable par rapport au scanner antérieur, avec une hypertrophie des glandes parotides et sous-maxillaires sans abcès collecté.
L’évolution est favorable après 6 jours d’antibiothérapie IV, avec une amélioration clinique franche et une nette diminution du syndrome inflammatoire (CRP à 45 mg/L). Les prélèvements bactériologiques (hémocultures) n’ont pas permis d’incriminer un germe spécifique. Un relais par amoxicilline/acide clavulanique 1 g 3 fois par jour par voie orale, dans l’hypothèse d’une infection des glandes salivaires à germes anaérobies, est proposé pour une durée de traitement de 15 jours au total.
Figure 2. Scanner cervical injecté (J0) montrant un aspect hypertrophié hétérogène des glandes parotides et sous-maxillaires avec dilatation canalaire.
• Sur le plan ophtalmologique
L’examen met en évidence une uvéite antérieure bilatérale non synéchiante, non hypertensive, non granulomateuse, associée à un syndrome sec oculaire au premier plan.
Les hypothèses diagnostiques avancées par l’équipe d’ophtamologie à ce stade sont : la sarcoïdose (bilan phosphocalcique, enzyme de conversion de l’angiotensine, scanner thoracique), la tuberculose (Quantiféron®), une maladie autoimmune (FAN et ANCA), l’arthrite juvénile idiopathique (HLA B27) et la maladie de Behçet (HLA B51). Un traitement par collyre corticoïde et larmes artificielles est mis en place et permet un contrôle progressif de l’uvéite et une diminution de la sensation de sècheresse oculaire.
Bilan étiologique
Un bilan étiologique à la recherche d’une étiologie infectieuse ou inflammatoire est réalisé. Le bilan hépatique et rénal est sans particularité. Il n’y a pas de protéinurie.
Le bilan phosphocalcique est normal et on ne note pas d’augmentation de l’excrétion urinaire du calcium. Le dosage pondéral des immunoglobulines IgG, IgA et IgM est normal. L’électrophorèse des protéines sériques est compatible avec un syndrome inflammatoire important (hyperalpha1, hyperalpha2).
On note également une diminution modérée de l’albumine et une hypogammaglobulinémie modérée à 7,7 g/L (n = 8-13,5).
Plusieurs hypothèses diagnostiques se posent donc : une cause infectieuse (notamment la tuberculose devant une altération de l’état général et la présence d’adénopathies cervicales, et l’herpès devant la présentation antérieure de l’uvéite), une maladie auto-inflammatoire ou auto-immune (sarcoïdose, maladie de Behçet devant la présence d’une uvéite non granulomateuse mais sans une atteinte postérieure qui est quasi constante dans le Behçet et l’absence d’autres atteintes cliniques telles qu’une aphtose, une pseudo-folliculite, des symptômes neurologiques ou des thromboses veineuses et/ou artérielles, etc.).
• Recherche d’une étiologie infectieuse
Le test Quantiféron® est revenu négatif. Sérologies Bartonella et rougeole négatives, sérologie des oreillons en faveur d’une immunité ancienne. Sérologie de Lyme, VIH, syphilis, toxoplasmose, HSV et CMV négatives, sérologie EBV en faveur d’une infection ancienne.
• Recherche d’une étiologie auto-immune ou auto-inflammatoire
Le bilan met en évidence : facteur rhumatoïde négatif, anticorps antinucléaires négatifs, anticorps anti-DNA natif négatif, ANCA négatifs, C3 et C4 normaux. HLAB27 et HLAB51 négatifs. ECA normale. Lysozyme à 15 mg/L (n = 3-12).
Une biopsie des glandes salivaires est finalement réalisée et révèle la présence de plusieurs granulomes sarcoïdosiques.
La tuberculose étant éliminée par le test Quantiféron® négatif, nous évoquons donc le diagnostic de sarcoïdose devant la présence de ces granulomes et, en particulier, le syndrome de Heerfordt qui associe classiquement une uvéite, une parotidomégalie, une paralysie faciale et de la fièvre.
Bilan d’extension de la maladie
Devant cette suspicion de sarcoïdose, un bilan d’extension est réalisé : scanner thoracique normal, EFR normales, échographies abdominale et rénale normales, ECG et échographie cardiaque normaux.
Le syndrome inflammatoire s’est normalisé à 3 semaines du début de la maladie (CRP négative, VS à 21 mm).
Prise en charge thérapeutique
Les symptômes de sécheresse buccale et oculaire sont au premier plan chez Ayoub. Un spray buccal Artisial® a été essayé mais sans amélioration sur les symptômes. Un traitement par corticothérapie per os en décroissance rapide sur un mois a été instauré (dose initiale à 1 mg/kg/j), compte tenu de l’efficacité avérée de la corticothérapie orale sur la plupart des manifestations cliniques de la sarcoïdose. L’évolution clinique à la fin de la corticothérapie a été favorable avec la disparition complète des symptômes de sécheresse buccale et oculaire, un meilleur état général et une reprise de poids.
La sarcoïdose chez l’adolescent
La sarcoïdose est une maladie systémique rare de cause inconnue.
Également appelée maladie de Besnier-Boeck-Schaumann, elle a été décrite pour la première fois par Besnier au XIXe siècle comme une maladie granulomateuse systémique touchant principalement les poumons et le système lymphatique. Sa physiopathologie est actuellement partiellement décrite. Il s’agit d’une dérégulation du système immunitaire liée à différents facteurs génétiques et environnementaux. La sarcoïdose est une maladie hétérogène concernant sa présentation clinique et son évolution. Il s’agit principalement d’une maladie de l’adulte, les enfants étant touchés environ 10 fois moins fréquemment.
Contrairement aux formes adultes de la maladie, les symptômes généraux sont souvent au premier plan lorsque le diagnostic est posé à l’âge pédiatrique et l’atteinte pulmonaire est alors exceptionnelle. Dans sa forme la plus courante, la sarcoïdose pédiatrique est une maladie multi-organes touchant préférentiellement l’oeil, la peau, les articulations, le système lymphatique et le foie, mais tous les organes peuvent être concernés. Une confirmation anatomopathologique après un examen rigoureux permettant d’exclure les diagnostics différentiels est indispensable chez les enfants chez qui la sarcoïdose reste un « diagnostic d’exclusion ». Elle se caractérise par une infiltration des organes touchés par des granulomes immunitaires épithélioïdes et gigantocellulaires sans nécrose caséeuse.
Ce diagnostic nécessite l’élimination des causes connues de granulomes, notamment le syndrome de Blau qui est une maladie monogénique granulomateuse exceptionnelle décrite chez les enfants en bas âge, la tuberculose et les autres infections à mycobactéries, les déficits immunitaires notamment le déficit immunitaire commun variable, la maladie de Crohn, les tumeurs et, plus rarement, la granulomatose médicamenteuse. L’incidence estimée de la sarcoïdose est de 0,6 à 1,02/100 000 enfants, mais en l’absence de registres internationaux, la maladie est probablement sous-déclarée.
Le syndrome de Heerfordt
Le syndrome de Heerfordt associe classiquement : uvéite, hypertrophie des glandes parotidiennes, paralysie faciale et fièvre modérée.
L’évolution est favorable dans la majorité des cas. Cette entité a été décrite pour la première fois par un ophtalmologiste danois Heerfordt en 1909. Waldenstrom en 1937 avait ensuite associé ce syndrome à la sarcoïdose. Le syndrome de Heerfordt est une entité rare et représente 4,1 à 5,6 % des patients atteints de sarcoïdose.
Classiquement, la « forme complète » est définie par les quatre symptômes présents (uvéite, hypertrophie de la glande parotide, paralysie faciale, fièvre) et les « formes incomplètes » par deux ou trois symptômes.
La stratégie thérapeutique du syndrome de Heerfordt n’a pas été établie en raison de la rareté de ce syndrome. Cependant, la majorité des cas de syndrome de Heerfordt décrits dans la littérature ont répondu à la corticothérapie orale de courte durée. Il n’y a pas de cas de corticodépendance décrit.
En conclusion
Nous rapportons un rare cas de syndrome de Heerfordt diagnostiqué chez un adolescent de 14 ans ayant présenté un tableau d’hypertrophie des parotides, associé à une sécheresse buccale et oculaire, et une uvéite. La présence de granulomes sarcoïdosiques retrouvés à la biopsie des glandes salivaires a permis de poser le diagnostic. La collaboration pluridisciplinaire est ainsi indispensable pour la prise en charge diagnostique puis thérapeutique des patients adolescents atteints de maladies systémiques telles que la sarcoïdose.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :