Publié le 10 fév 2017Lecture 5 min
Syndrome de Fahr et pseudohypoparathyroïdie
A. ES SEDDIKI*, A. AYYAD*, N. BENAJIBA*, H. HADJ KACEM**, *Service de pédiatrie, **Service de radiologie, CHU Mohamed VI, faculté de médecine et de pharmacie d’Oujda, université Mohammed 1er d'Oujda (Maroc)
La rubrique « En direct des staffs » est ouverte à tout médecin d’un service de pédiatrie souhaitant partager avec les lecteurs de Pédiatrie Pratique les cas discutés dans son service et qu’il estime suffisamment intéressants et édifiants pour être portés à la connaissance de ses confrères.
Défini par Theodor Fahr en 1930, le syndrome de Fahr (SF) est une entité anatomo-clinique rare, déterminée par la présence de calcifications intracérébrales, bilatérales et symétriques, non artériosclérotiques, localisées aux noyaux gris centraux(1). Habituellement asymptomatique dans les deux premières décennies, elle se manifeste vers l’âge de 30 ans par l’apparition de troubles neuropsychiatriques, ou vers l’âge de 60 ans par une démence progressive avec un syndrome extrapyramidal(2,3). La physiopathologie de cette affection reste mystérieuse(1-3). Le traitement de cette pathologie est basé sur la correction des troubles du métabolisme phosphocalcique, qui aboutit à une amélioration notable(1,2,4). Cette affection est habituellement associée à des troubles du métabolisme phosphocalcique, principalement à une hypoparathyroïdie, les autres dysparathyroïdies étant plus rares(2-4). Nous rapportons une observation de syndrome de Fahr ayant permis la découverte d’une pseudohypoparathyroïdie (PHP) chez un nourrisson de 9 mois.
Clinique
Il s’agit d’un nourrisson de sexe féminin, âgé de 9 mois de parents non consanguins, avec des antécédents de convulsion tonicoclonique généralisée apyrétique apparue deux mois auparavant, déjà mis sous valprote de sodium. Hospitalisé pour un état de mal épileptique évoluant depuis deux jours, le nourrisson gardait une hypotonie et une somnolence. L’examen clinique à l’admission montrait un poids à 7 kg (-2 DS), une taille à 70 cm (-1 DS), un périmètre crânien à 45 cm (normal), une tension artérielle correcte selon l’âge et une bandelette urinaire sans anomalie. L’examen neurologique montrait un nourrisson apyrétique, hypotonique et somnolent. Le fond d’œil était normal.
Les données biologiques montraient une hypocalcémie à 1,2 mmol/l,une hyperphosphorémie à 2,2 mmol/l, une hypocalciurie à 1,2 mmol/l, une hypophosphaturie à 8,79 mmol/l, un taux de phosphatases alcalines plasmatiques totales élevé à 512 UI/, un taux de thyréotropine plasmatique (TSH) normal à 1,66 mUI/l, un taux de thyroxine plasmatique (T4) normal à 15 pmol/ml, un taux de parathormone plasmatique (PTH) normal à 50 pg/ml et une hypovitaminose D à 18,5 ng/ml. Le reste du bilan biologique était normal. La sérologie de la toxoplasmose, de la rubéole et de l’HIV était négative.
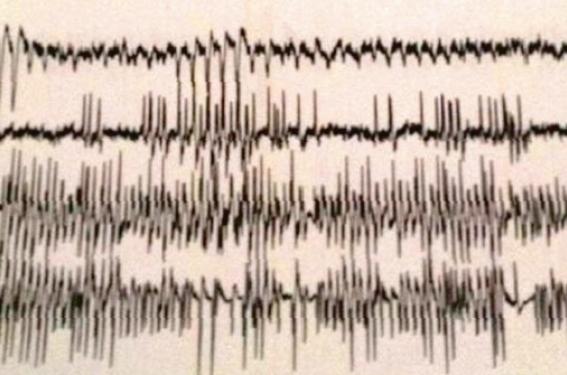
L’électroencéphalogramme a révélé une souffrance cérébrale diffuse et un foyer épileptique pariéto-temporal (figure 1). La radiographie du crâne de profil était normale, tout comme les radiographies du squelette. La tomodensitométrie cérébrale a montré de multiples calcifications péri-ventriculaires et des noyaux gris centraux de tailles variables réparties de façon bilatérale et symétrique (figure 2).
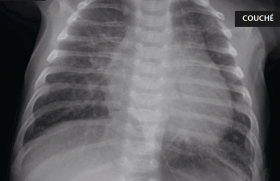
Figure 1. Électroencéphalogramme (EEG). Souffrance cérébrale diffuse
avec un foyer épileptique pariéto-temporale.
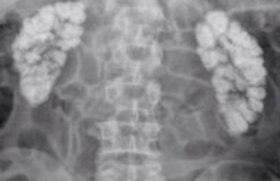
Figure 2. Tomodensitométrie cérébrale (TDM).
Multiples calcifications péri-ventriculaires et des noyaux gris centraux
de tailles variables réparties de façon bilatérale et symétrique.
L’imagerie par résonance magnétique cérébrale a objectivé des hyper signaux sur les séquences pondérées T2 touchant aussi les noyaux gris centraux. L’échographie cervicale était normale. Le diagnostic d’une pseudohypoparathyroïdie a été retenu pour notre nourrisson, révélée par un syndrome de Fahr. Un traitement substitutif a été instauré, associant calcium (2 g/j) et 1-alpha-hydroxy-vitamine D (1 mg/j), avec un traitement anticomitial (valproate de sodium). L’évolution a été marquée par la disparition des signes neurologiques et la normalité du bilan phosphocalcique.
Commentaires
Le syndrome de Fahr, défini par l’existence de calcifications cérébrales bilatérales et symétriques touchant les noyaux gris centraux, survient préférentiellement chez les patients présentant des dysparathyroïdies(1-3). Les mécanismes physiopatho logiques de la survenue des calcifications intracérébrales au cours du syndrome de Fahr sont mal élucidés (probablement pluri factoriels)(1,3-5). Les signes cliniques associés à ce syndrome sont divers: mouvements anormaux, signes psychiatriques, troubles cognitifs, dysarthrie, troubles de l’équilibre, ataxie, syndrome cérébelleux, signes pyramidaux, et crises d’épilepsie comme pour notre nourrisson(5-8). E. Turki et coll. ont rapporté dans une étude rétrospective tunisienne incluant dix patients suivis pour un SF dans un service de neurologie (dont l’âge moyen était de 27 ans), que les circonstances de découverte étaient des crises épileptiques chez cinq patients (50 % des cas), des troubles psychiatriques chez trois patients, des mouvements choréiques dans un cas et une paralysie faciale dans un autre(6). Ces calcifications cérébrales intéressent les petits vaisseaux des noyaux gris centraux.
La structure biochimique de ces calcifications comprend une matrice organique constituée de mucopolysaccharides et des éléments minéraux (calcium, phosphore, fer, magnésium, aluminium)(4,5). L’hypoparathyroïdie est la cause la plus classique de ce syndrome(2,3). Le diagnostic biologique est basé sur l’association caractéristique : une hypocalcémie, une hyperphosphorémie, une hypocalciurie, une hypophosphaturie, un taux élevé de PTH et une diminution du taux de la vitamine D. La pseudohypoparathyroïdie (PHP) réalise un tableau clinique et biologique similaire de l’hypoparathyroïdie, mais avec un taux normal de parathormone comme dans notre cas(3,4,5).
La revue de la littérature a permis de colliger une dizaine de cas SF associés à des hypoparathyroïdies, mais seulement deux cas de pseudo hypoparathyroïdie au cours du syndrome de Fahr ont été retrouvés chez l’enfant ; N. Kahloul et coll. ont rapporté un cas en 2009 chez un enfant tunisien de 12 ans, et Y. Sekkach et coll. ont rapporté un second cas en 2011 chez un enfant marocain de 12 ans(2,7). L’examen diagnostique de choix est la tomodensitométrie cérébrale, qui montre des calcifi cations bilatérales au niveau des noyaux caudés, lenticulaires, thalamiques, et parfois des noyaux dentelés.
En imagerie par résonance magnétique, les calcifications apparaissent en hyper signal sur les séquences pondérées T2(3,4,5,8). Certaines affections peuvent engendrer des calcifications intracérébrales (non bilatérales et non symétriques), notamment les maladies de système (lupus, sclérodermie), certaines infections (toxoplasmose, rubéole) et les tumeurs cérébrales primitives ou secondaires calcifiées(5,7,9). Par ailleurs, la maladie de Fahr (différente du SF) est une affection héréditaire, transmise sur le mode autosomique dominant par mutation du gène localisé sur le chromosome 14q, qui ne s’accompagne pas de troubles du métabolisme phosphocalcique(10). Sur le plan thérapeutique, la correction des perturbations du métabolisme phosphocalcique permet une disparition complète de l’ensemble de la symptomatologie neurologique et/ou neuro psychiatrique(3-6).
Conclusion
Notre observation souligne l’intérêt de rechercher des troubles du métabolisme phosphocalcique chez les patients porteurs de calcifications intracérébrales, et en particulier en cas de SF, pour pouvoir débuter un traitement approprié. Notre observation s’ajoute aux cas pédiatriques rares de pseudohypoparathyroïdie révélée par un SF.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :