Dermatologie
Publié le 18 jan 2018Lecture 6 min
Maladie de Von Recklinghausen chez l’enfant
Mouna KOURDA, Hôpital Razi, La Manouba, Tunisie
La neurofibromatose de type 1 (NF1) ou maladie de Von Recklinghausen représente 90 % des neurofibromatoses. Son incidence est estimée à 1/3 500. Elle est de transmission autosomique dominante, mais dans 50 % des cas, elle est sporadique. Elle est caractérisée par sa variation phénotypique au sein d’une même famille. La pénétrance du gène est complète après l’âge de 8 ans, âge où le tableau clinique s’enrichit des différentes manifestations cliniques. Le gène de la NF1 a été identifié, il est porté sur le chromosome 17(17q11.2). La neurofibromine est incriminée ; elle est à l’origine de la différenciation et de la prolifération cellulaire. Chez l’enfant de moins de 8 ans présentant des macules pigmentées isolées, se pose un double problème : celui du diagnostic positif des taches café au lait (TCL) et celui du diagnostic de NF1 devant des TCL isolées.
Les tâches café au lait
Les TCL sont fréquentes, présentes chez 2 % des nouveau-nés et 10 % des nouveau-nés de phototype foncé. Chez les adultes, elles sont rencontrées dans 10 à 20 % des cas, aussi bien chez les hommes que les femmes. Le plus souvent, elles sont physiologiques, isolées sans atteintes viscérales.
Les TCL typiques
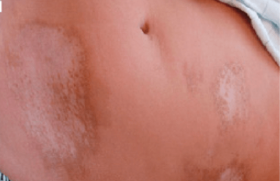
Au cours de la NF1, les TCL sont typiques, réalisant des macules, de couleur brun clair ; elles sont uniformes, à limites nettes et à contour variable. Leur forme est arrondie ou ovalaire, leur taille est variable, de quelques mm à quelques cm et de siège ubiquitaire (figure 1). Elles apparaissent précocement de la naissance à un an ; leur nombre et leur taille augmentent avec l’âge.
Les TCL atypiques
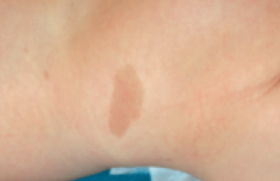
Elles sont caractérisées par leur grande taille et leur nombre est restreint (figure 2). Leur disposition est en bande ou blaschkolinéaire.
Au cours de la NF1, les TCL atypiques peuvent s’associer aux TCL typiques. De diagnostic clinique le plus souvent aisé, les TCL peuvent poser chez l’enfant, des problèmes de diagnostic différentiel avec de nombreuses pathologies (encadré ci-dessous).
Diagnostic ce NF1 en présence de TCL
Pour porter le diagnostic de NF1, 7 critères diagnostiques majeurs ont été précisés lors d’une conférence de consensus par le National Institute of Health (NIH) en 1988 (encadré ci-dessous). Pour porter le diagnostic de NF1, il faut au moins la présence de 2 critères majeurs sur 7. Par ailleurs, il est à souligner que les différentes manifestations cliniques cutanées et extracutanées sont de fréquence et d’âge d’apparition variable selon le type d’atteinte (encadré ci-dessous). Ceci est important à connaître afin d’en tenir compte lors de la demande d’explorations paracliniques et pour la conduite à tenir. Les TCL sont les manifestations cliniques les plus fréquentes et sont d’apparition précoce. Ainsi, chez les enfants, seules les TCL sont présentes et ce, durant de nombreuses années en l’absence d’antécédents familiaux. Se pose alors le problème diagnostique de NF1, car on est en présence d’un seul critère diagnostique. Il est à souligner que chez les enfants âgés de 8 ans, 5 % n’ont pas 2 critères pour confirmer le diagnostic de NF1.
Par ailleurs, d’autres syndromes, en plus de la NF1, peuvent comporter des TCL (tableau 1). Ainsi, le syndrome de Legius est apparenté à la NF1 ; son gène SPRED1 est différent de celui de la NF1. Ce syndrome comporte des TCL et des lentigines mais sans neurofibromes, ni nodules de Lisch, ni atteintes viscérales. Le syndrome de McCuneAlbright, dont le gène a été identifié GNA, comporte des TCL atypiques, de grande taille, à contour déchiqueté et de localisation systématisée.
Ce syndrome associe aux TCL une puberté précoce et une dysplasie fibreuse des os. Au cours de la NF1, d’autres manifestations cliniques, graves ou mineures, peuvent se voir (encadrés). Ainsi, les critères diagnostiques de la NIH doivent être révisés pour tenir compte de la richesse symptomatique de la NF1. Nous détaillerons certaines manifestations cliniques.
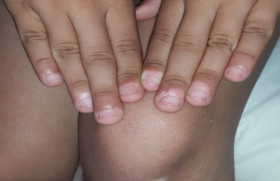
• Les lentigines Décrites par erreur comme des éphélides, les lentigines réalisent de petites macules de 1 à 3 mm. Elles sont pigmentées et considérées par certains auteurs comme des TCL de petite taille. De siège caractéristique aux plis axillaires et inguinaux, elles sont rares avant 2 ans et touchent 90 % des enfants âgés de 7 ans (figure 3).
• Les neurofibromes (NF) On distingue 3 types :
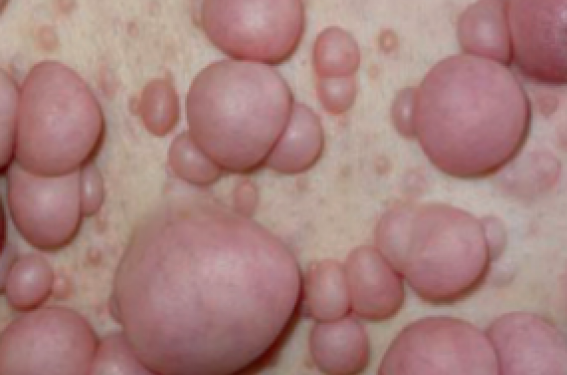
– les neurofibromes cutanés : ils apparaissent à la puberté et augmentent à la grossesse. Ils sont sensibles et douloureux (figure 4) ;
– les neurofibromes sous-cutanés : ils apparaissent à l’âge adulte, sont plus palpables que visibles et sont sensibles ou douloureux, pouvant entraîner des paresthésies (figure 5) ;
– les neurofibromes plexiformes, appelés aussi névromes plexiformes ou tumeurs royales : ils sont congénitaux ou apparaissent avant 5 ans. Il s’agit de tuméfactions cutanées et sous-cutanées, touchant un segment de membre (figure 6). La peau en regard est hypertrophique, pigmentée, pouvant présenter une hypertrichose. Ils sont profonds et l’infiltration touche la peau, les muscles et les viscères. Les neurofibromes plexiformes entraînent un préjudice esthétique très important.
• Les nodules de Lisch Ce sont des hamartomes iriens qui n’entraînent pas de troubles visuels (figure 7). Ils sont rencontrés dans 10 % des cas avant 6 ans et dans 90 % des cas après 16 ans. Les nodules de Lisch sont quasi pathognomoniques de la NF1. Ils sont visibles à la lampe à fente et parfois à l’œil nu.
• Le gliome des voies optiques (GVO) Il s’agit d’une tumeur cérébrale, rare mais grave, rencontrée dans 1,5 à 2 % des cas, et symptomatique dans la moitié des cas (figure 8).
• Les atteintes osseuses spécifiques : il s’agit de dysplasie osseuse, on en distingue 3 types selon la localisation :
– dysplasie des os longs : elle est congénitale touchant surtout le tibia. Les signes cliniques sont précoces à type de courbure de la jambe, spontanée ou lors de la marche. Les fractures et les pseudarthroses sont les principales complications (figure 9) ;
– dysplasie des ailes sphénoïdes : elle est congénitale et est associée à un neurofibrome plexiforme orbitaire. Rencontrée dans moins de 1 % des cas (figure 10), elle est grave, entraînant une asymétrie faciale, une exophtalmie et une énophtalmie ;
– dysplasie vertébrale : elle réalise une accentuation de la
concavité surtout postérieure ou antéro-postérieure des corps vertébraux, entraînant un aspect de feston (scalloping) avec élargissement des trous de conjugaison (figure 11). Les difficultés d’apprentissage constituent un problème majeur et sont retrouvées dans 30 à 40 % des cas. Ces troubles neurocognitifs altèrent la scolarité.
Il s’agit principalement d’un déficit de l’attention avec ou sans hyperactivité, des troubles de l’attention, des difficultés de la coordination motrice, d’un déficit de la mémoire récente et des difficultés d’élocution.
Conduite à tenir devant des TCL isolées en l’absence d’antécédents familiaux (figure 12)
Elle consiste :
– en un suivi clinique ;
– ou la demande d’une IRM cérébrale qui peut montrer des hypersignaux de la substance blanche en T2 dans 40 à 90 % des cas, dont des objets brillants non identifiés ou OBNI qui sont caractéristiques de la NF1 et régressent spontanément ;
– et/ou la demande d’un diagnostic moléculaire à la recherche du gène de la NF1. Un enfant qui présente plus de 6 TCL, de taille > 0,5 cm, sans antécédents familiaux, doit être considéré et suivi comme un patient atteint de NF1 jusqu’à preuve du contraire.
Évolutions et complications
La NF1 est une pathologie évolutive, caractérisée par la survenue de complications dans 15 à 20 % des cas, d’où l’intérêt de la surveillance. Les complications peuvent toucher différents organes (tableau 2).
NF1 et cancers
Au cours de la NF1, des cancers à type de tumeur maligne des gaines nerveuses sont rapportés. Ils apparaissent chez l’adulte et sont de pronostic catastrophique. Les facteurs de risque seraient la présence d’au moins 2 neurofibromes sous-cutanés. L’augmentation rapide d’un neurofibrome ou l’apparition de signes neurologiques déficitaires impose la biopsie chirurgicale urgente du neurofibrome. Il peut s’agir d’autres cancers : glioblastomes cérébraux, neuroblastomes, leucémies ou cancer du sein. La sévérité de la NF1 est variable et son évolution est imprévisible. La pratique d’examens paracliniques n’est pas rentable en l’absence de signes cliniques, à l’exception controversée de la demande de l’IRM cérébrale à la recherche d’un gliome des voies optiques chez des enfants de moins de 6 ans. Le suivi clinique se fait par une équipe multidisciplinaire impliquant le pédiatre et ultérieurement le généraliste et nous pensons qu’il devrait être effectué au moins une fois par an.
Conclusion
Devant des TCL isolées chez l’enfant dont le nombre est > 6 et la taille > 0,5 cm, on doit évoquer le diagnostic de NF1 et imposer un suivi clinique. En cas de confirmation ultérieure du diagnostic, lors de l’apparition des autres manifestations cliniques, la prise en charge doit être multidisciplinaire.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :