Publié le 10 fév 2011Lecture 14 min
Les grandes avancées des 50 dernières années en pédiatrie : le dépistage néonatal systématique
J.-P. FARRIAUX, Président Fondateur de l’Association régionale Nord-Pas-de-Calais de dépistage et de prévention des handicaps de l’enfant (ARDPHE)
Certes, le succès avec la phénylcétonurie, première encéphalopathie héréditaire évitée, a été universel et a suscité beaucoup d’espoir. Mais, peut-être a-t-on eu raison de parler de la « trop belle histoire de la PCU », car si on se place dans le cadre spécifique d’une action de santé publique, il existe un risque qu’une extension à tout va du programme de dépistage dégrade l’image formidablement positive de l’action actuelle, et que l’on n’ait plus la même efficacité, et ce pour un coût bien supérieur.
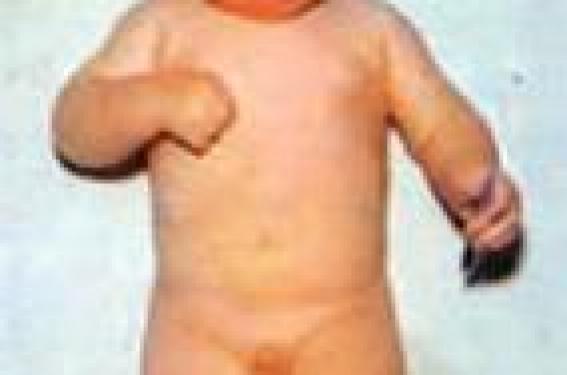
D’abord une définition « Le dépistage consiste à identifier présomptivement à l’aide de tests, d’examens ou d’autres techniques susceptibles d’une application rapide les sujets atteints d’une maladie ou d’une anomalie passée jusque-là inaperçue. Les tests de dépistage doivent permettre de faire le tri entre les personnes apparemment en bonne santé, mais qui sont probablement atteintes d’une maladie donnée, et celles qui en sont probablement exemptes. Ils n’ont pas pour objet de poser un diagnostic. Les personnes pour lesquelles les résultats sont positifs ou douteux doivent être envoyées à leur médecin pour diagnostic et, si besoin est, traitement » (1). Une belle histoire : la phénylcétonurie La phénylcétonurie est une anomalie héréditaire du métabolisme de la phénylalamine engendrant un retard psychomoteur majeur (figure 1). « La merveilleuse histoire de la phénylcétonurie » (2) et de sa prévention a été dans les années 70 le début d’une action originale de prévention secondairea. Figure 1. Phénylcétonurie. Initiée par des pédiatres en étroite collaboration avec les obstétriciens et les biochimistes, elle a permis « d’éradiquer » la phénylcétonurie (PCU) et de donner au malade porteur de cette anomalie, repérée en période néonatale, un développement psychomoteur normal grâce à un régime appauvri en phénylalanine. À son début, ce programme de prévention était un formidable pari, puisqu’il fallait utiliser des données dont on ne connaissait pas les règles précises. On savait simplement que : – la PCU était liée à un déficit en phénylalanine hydroxylase qui transforme la phénylalanine en tyrosine ; – le marqueur sanguin de la PCU, la phénylalanine (Phé), pouvait être dosé facilement à partir d’une goutte de sang recueilli sur papier filtre, selon la méthode microbactériologique décrite par Guthrie ; – la réduction de l’apport alimentaire en phénylalanine, rendue possible par la prescription d’un hydrolysat de protéines « débarrassées de la Phé », améliorait, comme l’avaient montré H. Bickel et coll. en 1954(3), l’état clinique des malades symptomatiques. Mais on ne savait pas : – si cette prescription diététique dans les premières semaines de vie permettrait effectivement un développement cérébral normal et la normalité ultérieure de l’individu ; – quel était le besoin minimum d’apport en phénylalanineb, acide aminé indispensable, et quel était en conséquence le taux acceptable de phénylalaninémie, et si ce régime « artificiel » n’aurait pas à longue échéance des effets néfastes majeurs notamment sur la croissance, voire a contrario sur le développement du cerveau luimême ; – s’il était envisageable et possible de réaliser un prélèvement de sang à tous les nouveau-nés sans exception ; en effet, le malade n’ayant aucun facteur de risque personnel ou familial permettant de le sélectionner a priori, c’est à toute la population qu’il fallait appliquer le test pour repérer le « suspect » dans la masse des nouveau-nés ; – si les médecins et les familles accepteraient ce prélèvement (au demeurant simple : piqûre au talon), et par la suite les conséquences induites par l’annonce d’un test positif (hyperphénylalaninémie), à savoir la prise en charge immédiate obligatoire pour vérification du diagnostic de PCU et prescription diététique stricte (pesage des aliments, etc.) à assurer pendant 8 à 10 ans ; – si la société, acceptant l’idée de ce dépistage systématique, voudrait bien en assurer le financement, en sachant que si le test unitaire est peu onéreux (± 1,7 €), sa réalisation à l’échelle d’une nation et pour les décennies à venir entraînerait une dépense très importante, sans omettre par ailleurs que le coût des produits diététiques spécifiques était très onéreuxc. Ainsi, le défi lancé, plus sur des hypothèses que des certitudes, était pour le moins risqué. Et ce qui a été « merveilleux », c’est qu’il a été gagné, puisque aujourd’hui tous les nouveau-nés (± 99,9 %) ont un test de Guthrie et que les PCU sont bien repérées. Traités, les sujets dépistés sont tous devenus des adultes normaux. Épopée de la mise en place d’un programme national de dépistage néonatal systématique Cela n’a pas été simple à organiser. Il a fallu la bonne volonté de tous (médecins, sages-femmes…), la mise en place d’un organigramme efficace (figure 2) par la Fédération nationale (AFDPHE) des associations régionales (une par région administrative), chacune ayant la totale responsabilité de la réalisation du programme dans son territoire. Figure 2. Organigramme du programme français de dépistage néonatal systématique. Il a fallu obtenir le soutien financier de la CNAMTS, qui a bien voulu participer à la réalisation de l’action dans le cadre d’un partenariat efficace et pérenne avec l’AFDPHE, en même temps que le ministère de la Santé acceptait de créer une nouvelle classe de produits remboursables par la Sécurité sociale, les éléments médicaments, et qui fut un précieux apport pour les parents. À la fin 2009, 30 millions de nouveau- nés français ont eu un test de Guthrie et 1 725 sujets PCU ont été repérés et traités. Pendant toutes ces décennies, nous avons tous appris à manier le programme, les malades et leur traitement. Il a fallu aussi nous familiariser avec des données nouvelles, comme par exemple les hyperphénylalaninémies malignes par anomalies du système co-factoriel bioptérine, qui ne réagissent pas au régime PCU, ou le risque de foetopathologie sévère chez le « nouveau-né d’une mère PCU », risquant d’annuler à la 2e génération le bénéfice obtenu à la 1re avec le traitement préventif de la mère. Concernant ce risque, parmi les approches possibles, la plus pertinente a été retenue, à savoir la reprise préconceptionnelle du régime PCU chez toute femme PCU souhaitant un enfant ; là encore, le pari était risqué, mais nous savons aujourd’hui qu’il est gagné. Les mères PCU ont des enfants normaux. Nous avons aussi appris à assimiler les critères précis devant régir tout programme de dépistage systématique, tels que les avaient édictés J.M.G. Wilson et G. Jungner sous l’égide de l’OMS (4), et qui n’ont pas été à ce jour réellement remis en cause (tableau 1). Et dans ce désir compréhensible d’étendre le programme à d’autres affections, nous avons dû tenir compte de multiples éléments – faisabilité, fiabilité, sensibilité et spécificité, acceptabilité, ethnicité, utilité et rendement (coût/bénéfice)…–, dont la conjonction positive peut justifier l’inclusion d’une affection dans le programme. Les autres maladies dépistées L’extension du dépistage néonatal systémique a donc été prudente, ne s’adressant qu’à des maladies répondant strictement aux critères de Wilson. ● L’hypothyroïdie congénitale, dont tous les pédiatres connaissaient plus ou moins bien la symptomatologie (figure 3) et dont ils savaient que le traitement au stade clinique n’évitait pas toutes les séquelles, notamment cérébrales. Le programme a été initié en 1978. Plus de 24 millions d’enfants ont été testés et 7 091 malades repérés avec les formidables résultats que l’on connaît. ● L’hyperplasie congénitale des surrénales est aussi une maladie qui cadrait bien avec les critères du dépistage, et notamment l’efficacité du traitement sur la létalité précoce du syndrome de déshydratation avec perte de sel et aussi la redoutable erreur d’assignation de sexe. Depuis 1995, 13 millions de nouveaunés ont été testés et 702 malades repérés avec d’excellents résultats de la prise en charge néonatale. Elle est toutefois peu connue par certains étant donné la relative rareté de la maladie (1/19 000) et le peu de fiabilité du test 17OHP chez le prématuré, qui présente très souvent une hyper-17-OHprogestéronémie transitoire. Figure 3. Hypothyroïdie congénitale. ● La drépanocytose (HbS). L’approche était justifiée par la gravité des épisodes infectieux, souvent létaux dans les premières années de vie, et l’efficacité d’une thérapeutique préventive par antibiothérapie et vaccination systématique. L’obstacle était qu’en pratique le syndrome drépanocytaire ne touchant que quelques populations à risque, il ne paraissait pas rationnel de pratiquer un test chez tous les nouveau-nés, alors qu’un quart seulement sont issus de familles à risque (Afrique Noire essentiellement). On s’est donc résolu à un dépistage « ciblé » en Métropole, basé sur des signes de distinction raciale, et systématique dans les régions à risque (Antilles, Guyane et Réunion). Plus de 3 millions de nouveau-nés ont été testés avec 4 656 malades identifiés et une réduction drastique du taux de mortalité dans le jeune âge. Deux interrogations demeurent cependant, l’une à propos du repérage des sujets hétérozygotes et normaux (ou presque) et de l’utilisation de ce résultatd, et l’autre sur l’avenir du malade qui ne tire pas de bénéfice dans l’évolution lointaine de son affection. ● La mucoviscidose a été ajoutée au programme en 2002, étant donné sa fréquence (1/3 500), sa gravité et la disposition d’un dépistage efficace avec le couplage trypsine immunoréactif + génotypage. Il a permis de repérer 1 341 malades (sur près de 6 millions de nouveau-nés testés). Ces enfants ont tous bénéficié d’une prise en charge multidisciplinaire précise dans des unités spécialisées (Centre de ressource et de compétence de la mucoviscidose, CRCM), ce qui allie l’avantage d’un diagnostic précoce et d’une prise en charge adaptée. Toutefois, si dans le jeune âge il y a des bénéfices évidents sur le plan nutritionnel, il n’est pas sûr que l’évolution à long terme soit radicalement modifiée, la durée moyenne de vie n’étant pas allongée de façon évidente qu’il y ait eu ou non un dépistage néonatal. Comme pour l’HbS, on repère aussi des hétérozygotes porteurs sains qui ont fait l’objet d’un riche argumentaire dans l’avis du CCNEd. ● D’autres maladies ont été testées (tyrosine, déficit en désaminases, neuroblastome, maladie de Duchenne, galactosémie, déficit en G6PD, etc.), mais elles n’ont pas résisté à une expérimentation très stricte. Un programme expérimental de dépistage de la surdité à la naissance est actuellement en cours (DGS – CNAMTS – AFDPHE) ; il confirme la faisabilité du dépistage en maternité et son efficacité (repérage des enfants, acquisition des outils de communication quasi normale). Bilan et devenir Le bilan de l’action de dépistage néonatal systématique est globalement très positif, et notamment pour la phénylcétonurie et l’hypothyroïdie congénitale. Pour les autres affections, les résultats sont moins probants, ce qui oblige à se poser des questions sur l’utilité du dépistage de telle ou telle autre maladie pour peu qu’elle soit dépistable. Les moyens techniques mis à notre disposition sont très évolutifs, et nous pouvons déjà à partir d’une seule tache de sang dépister une trentaine de maladies héréditaires du métabolisme. L’utilisation du génotypage devrait décupler le nombre des maladies dépistables. Mais estce utile et réellement efficace ? Est-ce souhaitable et acceptable ? La demande est « pressante » et nous invite à réaliser beaucoup plus de tests d’affections dépistables. Les intervenants sont ici multiples : • les médecins, avec les pédiatres, y sont relativement favorables, mais de façon pondérée. Ils savent que l’annonce du diagnostic aux parents est plus aisée lorsqu’il existe un traitement éprouvé de la maladie, et beaucoup plus difficile et mal ressenti s’il n’y a pas vraiment de projet thérapeutique efficace. Les spécialistes des maladies métaboliques sont plus favorables à un multidépistage, arguant que c’est un moyen d’étudier le développement naturel de la maladie, mais aussi de tester de nouvelles thérapeutiques ; • les biologistes sont assez favorables à un dépistage général, car il leur permet d’approfondir les recherches biochimiques et d’utiliser de nouvelles technologies (double spectrométrie de masse, puce à ADN, etc.) ; cependant, ils reconnaissent qu’ils risquent d’être confrontés à des situations difficiles à résoudre – « profil » biologique anormal mais non classable ; anomalie bien identifiable mais quantitativement modérée (s’agit-il alors d’un « vrai » malade ou d’un variant « modéré ») ; il n’y a pas toujours une liaison évidente entre anomalies biologiques et maladie clinique ; • les parents peuvent avoir des positions divergentes : – certains veulent savoir pour éviter une quête médicale parfois longue et toujours éprouvante ; – d’autres pensent qu’un diagnostic, même lorsqu’il n’existe pas de traitement, pourra leur permettre un autre projet de grossesse en maîtrisant le risque reconnu (par exemple : diagnostic prénatal ou insémination avec donneur…) ; – et il y a aussi ceux qui préfèrent ne pas savoir et profiter de leur bébé tant qu’il va bien. Il est évident que dans le cadre d’un programme de dépistage systématique, leur consentement sera demandé, et que les parents sont en droit de refuser le test. Mais, peut-on a priori être sûr qu’ils auront tous reçu une information claire, précise et compréhensible, et qu’ils ne se trouveront pas dans la situation dramatique d’avoir à vivre avec un bébé temporairement normal et atteint d’une maladie grave non traitable ? De nombreux autres groupes peuvent aussi intervenir : éthiciens, sociologues, économistes de la santé, spécialistes de santé publique, représentants religieux, etc., et ce n’est qu’avec leur consensus que l’on pourra élargir le panel des maladies à dépister. Entre ne pas dépister et dépister tout ce qui est possible, il y a certainement un juste milieu à trouver. Lorsque l’on poursuit réellement un objectif de santé publique, il y a des règles à respecter et, aux critères de Wilson, on peut ajouter la réflexion proposée par J.-P. Brodin et M. Boissel (5) qui ont listé les items à examiner avant la mise en route d’un dépistage (tableau 2). Nous encourageons le lecteur intéressé à consulter l’article de W. Dab, qui propose une démarche prudente pour aboutir à des choix rationnels de dépistage (6). Conclusion Le programme national de dépistage néonatal systématique est une nouvelle donne dans la médecine préventive. Les pédiatres ont vite compris son intérêt et la population l’accepte avec facilité par un simple consentement tacite. Les textes récents (arrêté du 22 janvier 2010 fixant la liste des maladies donnant lieu à un dépistage néonatal et en application de l’article R.1131- 21 du code de la Santé publique) en officialisent la prescription (tableau 3).
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :