Publié le 25 nov 2024Lecture 4 min
Hypochromie, yeux bleus et surdité
Asmaa LAHROUGUI*, Cheick AK WAGALO**, Dalila BENLAHCENE***, Emmanuel MAHÉ* - *Service de dermatologie, hôpital Victor Dupouy, Argenteuil **Centre de santé de référence de Koutiala, Mali ***Cabinet libéral, ACPP, Casablanca, Maroc
Le service de dermatologie d’Argenteuil, dans le cadre d’une collaboration internationale soutenue par l’Association casablancaise des pédiatres privés (ACPP), a ouvert début 2024 une solution de téléexpertise l’unissant à 248 pédiatres francophones de 7 pays : Maroc, Algérie, Sénégal, Tunisie, Mali, Niger, et Burkina Faso(1). Cette observation illustre l’intérêt de cette solution pour des cas complexes, ici adressée par un collègue malien.
Cas clinique
Une jeune fille âgée de 10 ans, de Koutiala (Mali) présente depuis la naissance des troubles pigmentaires associés à une surdité de perception et une mutité. L’interrogatoire révèle une symptomatologie similaire familiale, notamment chez la mère et les deux sœurs. Il n’y avait pas de notion de consanguinité.
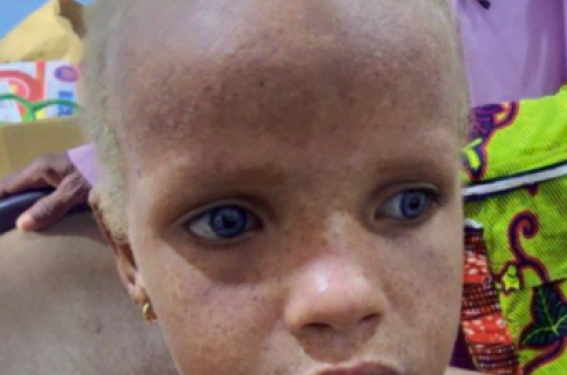
Les photos adressées montrent des cheveux clairs, des iris bleu saphir, ainsi que la présence de macules achromiques en confetti au niveau du visage, du tronc et de l’abdomen (figure 1).
Figure 1. Yeux bleus, cheveux clairs, hypopigmentation mouchetée diffuse.
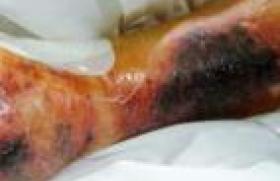
On note également la présence d’un placard achromique segmentaire du membre inférieur droit touchant la cuisse et la face antéropostérieure de la jambe droite (figure 2), ainsi qu’un élargissement de la racine du nez (figure 1).
Figure 2. Grande plaque vitiligoïde de la jambe.
L’aspect clinique et le caractère familial sont en faveur du syndrome de Waardenburg. Sa prise en charge repose sur la réalisation d’un conseil génétique de la patien te et de sa famille (dans la mesure du possible), une photoprotection adaptée, une évaluation neurosensorielle et auditive, ainsi qu’un suivi psychologique.
Le syndrome de Waardenburg
Le syndrome de Waardenburg (WS) est un groupe de maladies génétiques rares transmis selon un mode autosomique dominant, décrit en 1951 par l’ophtalmologiste et généticien néerlandais, Petrus Johannes Waardenburg. Sa prévalence mondiale est estimée à 1/42 000 individus, avec une égalité racia le et du sexe ratio.
Il existe plusieurs types de SW. Dans le type I, le diagnostic est clinique, associant deux critères majeurs ou un critère majeur et deux critères mineurs.
• Critères majeurs :
– synophris (implantation pilaire de la région intersourcilière) ;
– surdité (30 à 50 %) neurosensorielle de sévérité variable ;
– dystopie des canthi ;
– hétérochromie (yeux vairons), coloration bleue des iris ;
– mèche blanche frontale ou blanchissement précoce des cheveux avant l’âge de 20 ans.
• Critères mineurs :
– leucodermie congénitale ;
– racine du nez large ;
– hypoplasie des ailes du nez.
Toutefois, il existe un polymorphisme clinique et une disparité génétique scindant ce syndrome en quatre sous-types, avec une prédominence du type I et II :
– le WS de type I est dû à des mutations du gène PAX3, caractérisé par une surdité de perception, une dystopie du canthorum, une racine nasale large, un philtrum court et un maxillaire rétropositionnel court ;
– le WS de type II est due à des mutations du gène MITF avec des caractéristiques semblables au type I, hormis une distance normale des canthi internes des deux yeux ;
– le WS de type III (syndrome de Klein-Waardenburg) se distingue par des anomalies des membres supérieurs, notamment une hypoplasie du système musculosquelettique, des contractures en flexion, des fusions des os du carpe, des syndactylies ;
– le WS de type IV, également appelé syndrome de ShahWaardenburg, est due à des mutations auto somiques récessives de l’EDNRB ou de l’endothéline-3. Il présente les mêmes caractéristiques cliniques que le type II, avec une association au mégacôlon congénital (maladie de Hirschsprung).
La surdité est due à des mutations des gènes MITF et PAX3 ridentifiés dans les types I et II, associée dans certains cas à une atrophie du ganglion spinal, une réduction des fibres nerveuses et une absence de l’organe de Corti.
Le diagnostic différentiel se pose principalement avec le piébaldisme, le syndrome de Tietz, l’albinisme oculocutané, la maladie de Vogt-Koyanagi-Harada et le vitiligo.
L’espérance de vie des patients est normale, néanmoins les complications sont liées aux anomalies des tissus dérivés de la crête neurale et spécifiques à certains variants du syndrome de Waardenburg, notamment le blépharophimosis pour le type I, le handicap mental et la microcéphalie pour le type III, ainsi que la maladie de Hirschsprung pour le type IV. Il n’existe aucun traitement curatif pour le syndrome de Waardenburg, la prise en charge est pluridisciplinaire et symptomatique visant à améliorer la qualité de vie des patients par un soutien psychologique, à traiter les comorbidités et les complications par des implants cochléaires, ainsi qu’à appliquer une photoprotection adaptée préventive.
La sensibilisation des patients et leur entourage à la réalisation d’un conseil génétique est importante, permettant de prédire le risque de transmission générationnelle, sans prédiction de la gravité de son expression clinique.
Les troubles pigmentaires et auditifs chez l’enfant peuvent avoir des répercussions sociales et psychologiques conséquentes, d’où l’importance d’un diagnostic et d’un traitement précoces, associés à un suivi psychologique au long cours.
Pour en savoir plus :
• Mahé E. Télédermatologie en pédiatrie, une optimisation du parcours de soins ? Réalités Pédiatriques 2024 (sous presse).
• Orpahnet. Syndrome de Waardenburg types 1, 2, 3 ,4 ; Disponible sur https:// www.orpha.net/pdfs/data/patho/Han/Int/fr/ SyndromeDeWaardenburg_FR_fr_HAN_ ORPHA3440.pdf.
• Ilyaz M, Jadhav R S, Pande V, et al. Waardenburg Syndrome: A Report of a Rare Case. Cureus 2024 ; 16(7) : e65704.
• Yu Y, Liu W, Chen M, et al. Two novel mutations of PAX3 and SOX10 were characterized as genetic causes of Waardenburg Syndrome. Mol Genet Genomic Med 2020 ; 8(5) : e1217.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :