Pédiatrie générale
Publié le 01 fév 2009Lecture 37 min
Syndromes myasthéniques congénitaux : expression phénotypique et caractérisation physiopathologique
B.EYMARD, Centre de Référence de Pathologie Neuromusculaire Paris-Est, Institut de Myologie
Les syndromes myasthéniques congénitaux (SMC) constituent un groupe d’affections génétiques à l’origine d’un dysfonctionnement de la transmission neuromusculaire, beaucoup plus rares que les myasthénies auto-immunes. Cependant la fréquence des SMC est probablement sous estimée du fait de nombreux cas non diagnostiqués. Au cours des 20 dernières années, des progrès remarquables ont été faits dans l’épidémiologie, la caractérisation phénotypique, le diagnostic moléculaire et le traitement des SMC. Pour autant les gènes impliqués n’ont été identifiés que dans la moitié des formes de SMC. C’est dire qu’il reste beaucoup de travail à accomplir, qui réclamera la collaboration internationale des morphologistes, généticiens et neurobiologistes.
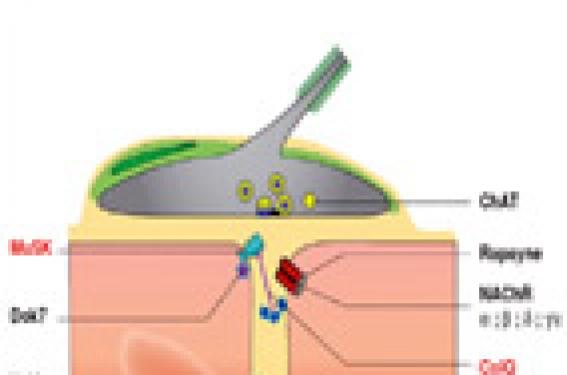
Introduction Les syndromes myasthéniques congénitaux (SMC) constituent un groupe hétérogène d’affections génétiques responsables d’un dysfonctionnement de la transmission neuromusculaire qui se manifeste par une faiblesse musculaire accentuée par l’effort, débutant le plus souvent dans la petite enfance. Leur pathogénie les oppose aux syndromes myasthéniques acquis d’origine auto-immune, représentés par la myasthenia gravis et le syndrome présynaptique de Lambert-Eaton, respectivement dus à des anticorps dirigés contre le récepteur de l’acétylcholine (RACh) et les canaux calciques de la terminaison nerveuse. Les SMC sont beaucoup plus rares que la myasthénie auto-immune. Leur fréquence est estimée à 1:500000 en Europe. Ces chiffres sont extrapolés à partir du nombre des syndromes myasthéniques débutant dans la petite enfance (1 à 2 %), dont l’origine congénitale est hautement probable (Millichap et al., 1960), mais ils sont probablement sous estimés car le diagnostic de SMC n’est pas toujours posé (voir infra). Le développement des connaissances concernant les SMC s’est considérablement accéléré dans les 30 dernières années, à la suite de nombreux travaux menés par le groupe d’A Engel. En 1977, le déficit en acétylcholinestérase (AChE) fut le premier SMC caractérisé sur le plan physiopathologique : l’enzyme était absent au niveau des jonctions neuromusculaires (Engel et al., 1977). Progressivement l’hétérogénéité physiopathologique des SMC a été établie : à côté du SMC synaptique dû au déficit en AChE, d’autres SMC ont été identifiés, correspondant à des anomalies de molécules situées dans la région postsynaptique (versant musculaire de la jonction neuromusculaire) et dans la région présynaptique (terminaison du nerf moteur). Dès le début des années 80, l’analyse microélectrophysiologique de la transmission neuromusculaire, effectuée sur muscle intercostal, a permis d’identifier le syndrome du canal lent (slow channel syndrome), correspondant à une anomalie cinétique du récepteur de l’acétylcholine (RACh) dont l’ouverture était anormalement prolongée (Engel et al.,1982). Dans les 20 dernières années, grâce aux progrès remarquables de la génétique moléculaire, 10 gènes ont été identifiés dont le dernier, DOK7, en 2006 (Beeson et al., 2006) (figure). Parmi les SMC caractérisés, environ 85 % sont postsynaptiques, avec 3 gènes principaux impliqués : le gène de la sous-unité epsilon du RACh, de la rapsyne et de Dok7 ; d’autres gènes sont plus rarement en cause : ceux des sous-unités α, β,δ du RACh, du canal sodium (SCN4A), et d’une tyrosine kinase spécifique du muscle (MuSK). Dix pour cent des SMC sont synaptiques et 5 % présynaptiques, avec dans les 2 cas un seul gène identifié codant respectivement pour la chaîne collagénique de l’acétylcholinestérase (Colq) et pour la choline-acétyltransférase (ChAT). L’expression des mutations dans des modèles cellulaires et des modèles de souris invalidées pour le gène ou porteuses du gène muté ont permis de prouver et de comprendre le rôle pathogène des mutations. En dépit de ces avancées remarquables, 50 % des SMC restent non caractérisés sur le plan moléculaire. De nombreuses revues ont été consacrées aux SMC, dont les plus récentes en 2005 et 2007 (Beeson et al., 2005; Müller et al., 2007). L’objectif de cet article est de présenter les principales caractéristiques phénotypiques et physiopathologiques des SMC et de proposer une stratégie pour la caractérisation précise de ces affections. Expression clinique des SMC Les différents SMC, quelle que soit leur physiopathologie, partagent une présentation clinique commune. Le début est en général très précoce, typiquement dès la période néonatale ou lors des première années de vie. Les acquisitions motrices sont souvent retardées. Une arthrogrypose, un hydramnios sont plus rares, témoignant d’un début fœtal. L’apparition tardive des symptômes à l’adolescence, voire à l’âge adulte, a été plus rarement rapportée. Certains signes cliniques orientent vers une anomalie de la transmission neuromusculaire : ophtalmoplégie et ptosis, signes bulbaires (dysphonie, troubles de déglutition), parésie faciale, fatigabilité musculaire. Chez le jeune enfant, le ptosis est d’appréciation délicate, l’hypotonie, la pauvreté de la mimique, les troubles de succion, la faiblesse du cri sont au premier plan. La survenue de poussées est caractéristique, tout comme l’aggravation par l’effort. L’effet favorable des anticholinestérasiques, qu’ils soient administrés par voie parentérale au cours d’un test à la prostigmineR ou par voie orale (souvent préférée chez le jeune enfant), est un argument important en faveur du syndrome myasthénique; néanmoins certains SMC sont aggravés par les anticholinestérasiques : le syndrome du canal lent , le déficit en AChE et en DoK7 (voir infra). Aux signes proprement myasthéniques, s’associent fréquemment des éléments myopathiques : amyotrophie, scoliose, rétractions. Une lenteur de la réponse à la lumière est propre au SMC dû à un déficit en AChE. La gravité des SMC est très variable, dépendant de l’intensité du déficit moteur, des troubles bulbaires et de l’atteinte respiratoire. La survenue d’épisodes de défaillance respiratoire très aiguë, favorisée par des épisodes infectieux, est fréquente dès les premiers mois de vie. Annoncée par un encombrement soudain, la poussée respiratoire se traduit par une bradycardie, des apnées. En l’absence d’assistance respiratoire, le risque vital est majeur. Ces épisodes apnéiques sont fréquents dans les SMC dus à une anomalie primaire de ChAT et de la rapsyne. L’existence d’une histoire familiale est un argument essentiel en faveur du caractère génétique du syndrome myasthénique. La majorité des SMC est de transmission autosomique récessive, ce qui explique la fréquence des cas sporadiques dans les petites fratries. Le syndrome du canal lent est le seul SMC autosomique dominant caractérisé jusqu'alors. Les profils évolutifs des SMC sont très variés, y compris chez un même patient, à différentes périodes de la maladie. Les poussées myasthéniques se manifestent par une aggravation rapide de la faiblesse ou de la fatigabilité des membres, des troubles oculo-bulbaires, une décompensation respiratoire. Elles surviennent à tous les âges et leur durée est très variable, de quelques jours à plusieurs années. Des facteurs déclenchants sont souvent retrouvés : épisodes infectieux, grossesse. Une aggravation progressive de la maladie est fréquente, survenant parfois tardivement à l’âge adulte avec déficit moteur conduisant au fauteuil roulant, apparition d'une insuffisance respiratoire motivant une ventilation assistée. Une évolution favorable spontanée ou favorisée par l'instauration du traitement peut survenir après un début néonatal sévère. Les thérapeutiques facilitatrices de la transmission neuromusculaire ont permis chez certains patients d’améliorer le pronostic. Description des syndromes myasthéniques congénitaux Les SMC postsynaptiques sont de loin les plus fréquents (environ les trois quarts des formes identifiées). Par ordre de fréquence décroissante, dans l’expérience de notre réseau national de SMC, les gènes impliqués sont sous-unité α du RACh (45 %) > Rapsyne (19 %) > Dok7 = ColQ (14 %) >> MuSK (3 %) > sous-unité ε du RACh (2 %) > sous-unité β et δ du RACh (1 %), > SCN4A (0 %). Parmi les SMC dus à une anomalie primaire du RACh, 2 catégories sont distinguées, ceux avec anomalie cinétique, ceux plus nombreux avec perte en RACh, sans anomalie cinétique. SMC avec anomalies cinétique du RACh Le syndrome du canal lent ou « slow channel syndrome ». Il s’agit de l’anomalie cinétique du récepteur la plus fréquente. Cette entité, de transmission autosomique dominante, a été décrite dès 1982 (Engel et al., 1982). Elle est caractérisée par un allongement du temps d’ouverture du RACh que révèle l’étude microélectrophysiologique sur muscle intercostal (Engel et al., 1982). Une vingtaine de mutations ponctuelles faux sens du RACh entraînant un gain de fonction à effet dominant ont été identifiées (pour revue, Engel et al., 2005). Dans la plupart des cas, l’allongement du temps d’ouverture était démontré par la technique de patch clamp qui permet l’enregistrement unitaire du RACh porteur de la mutation exprimé dans la cellule HEK. Les mutations les plus fréquentes intéressent la sous-unité α (Sine et al., 1995 ; Engel et al., 1996a ; Milone et al., 1997 ; Ohno et al., 2000a) mais les autres sous-unités peuvent être impliquées (Ohno et al., 1995a ; Ohno et al., 1998a). Le siège des mutations intéresse : 1) deux domaines transmembranaires participant à la formation du pore au travers duquel passe le flux sodique (Wang et al., 1997) : M1, pour les mutations de la chaîne α et β (Ohno et al., 2000a) et M2, pour celles, plus fréquentes, affectant les chaînes α, β, δ, ε ; 2) une région du domaine extracellulaire de la sous-unité α au voisinage du site de fixation de l’ACh (mutations αG153S et αV156M). Les conséquences fonctionnelles des diverses mutations ont été étudiées in vivo sur biopsie de muscle intercostal ou in vitro en exprimant la mutation dans des systèmes cellulaires (Croxen et al., 1997). L’allongement de la durée d’ouverture du RACh est lié soit à une lenteur de la fermeture du canal, en cas de mutations situées dans la région du pore, soit à une augmentation de l’affinité du récepteur pour son ligand pour les mutations du domaine extracellulaire (Engel et al., 1996a). Dans les 2 cas, il existe un gain de fonction du RACh. Une observation de syndrome du canal lent avec à la fois un temps de fermeture prolongé et une ouverture retardée, dû à une mutation de la sous-unité δ du RACh a été rapportée (Gomez et al. 2002). En 2002, Croxen et collaborateurs ont publié une observation très particulière de syndrome du canal lent de transmission récessive, survenant dans une famille consanguine, en rapport avec une mutation homozygote de la sous-unité ε (εL78P), située dans la région extramembranaire. Cette mutation n’était pathogène que si elle était présente sur les 2 allèles (Croxen et al., 2002a). Par ailleurs, un syndrome du canal lent associé à une translocation chromosomique 2q31-9p27 a été décrit récemment (Zeevaert et al., 2002). L’expression clinique est variée: certains cas sont précoces et sévères, d’autres tardifs, avec un début autour de 20 ans et une atteinte modérée (Engel et al., 1982 ; Oosterhuis et al., 1987). Trois arguments orientent vers un syndrome du canal lent : l’hérédité autosomique dominante, l’absence de réponse aux anticholinestérasiques, le dédoublement du potentiel moteur après une stimulation unique. La sélectivité de la formule clinique est aussi évocatrice, comportant un déficit atrophiant prédominant sur les extenseurs des doigts des mains et les muscles cervicaux. Des remaniements importants de structure de la plaque motrice sont retrouvés en microscopie électronique avec dépôts de calcium, destruction majeure des replis postsynaptiques, vacuolisations, agrégats tubulaires (Engel et al., 1982). La caractérisation précise de ce syndrome est à l’origine d’un traitement étiopathogénique par la quinidine, un agent bloqueur capable de normaliser la durée d’ouverture du RACh (Fukudome et al.1998; Harper et Engel, 1998). La fluoxetine, à la dose de 80 à 120 mg a également un effet favorable (Harper et al. , 2003). Le syndrome myasthénique du canal rapide (fast channel syndrome). Ce syndrome également rapporté par le groupe d’Engel est de transmission autosomique récessive. A la différence du syndrome du canal lent, aucun élément clinique ou électromyographique n’est spécifique au syndrome du canal rapide. Sa reconnaissance passe par l’étude microélectrophysiologique sur muscle intercostal ou sur cellules HEK porteuse des mutations qui révèle un raccourcissement du temps d’ouverture du RACh, anomalie en miroir du syndrome du canal lent (Uchitel et al., 1993 ; Sine et al., 2003). La sévérité clinique est variable. Une arthrogrypose a été rapportée dans un cas (Bronlow et al., 2001). Les patients répondent à la combinaison 3,4-DAP - anticholinestérasiques. Huit mutations ont été identifiées affectant les sous unité α, δ, ε situées soit dans la région extracellulaire, soit au niveau du domaine membranaire M3 (mutation alpha V285I) soit dans la boucle cytoplasmique comprise entre les domaines M3 et M4 (mutations ε uniquement) (voir pour revue Engel et al., 2005). Les syndromes myasthéniques avec déficit en RACh prédominant (anomalies cinétiques absentes ou minimes) Ils représentent 40 à 50 % des cas de SMC identifiés (Engel et al., 1996b). Il existe un effet fondateur dans la population gitane : mutation homozygote ε 1267delG (Abicht et al., 1999; Hercegfalvi. et al., 2000) et dans la population du Maghreb : ε ins1293 G homozygote. A côté de ces mutations ethniques, les mutations décrites sont très nombreuses (une soixantaine) soit homozygotes soit hétérozygotes (pour revue, Engel et al., 2005). Elles sont de tous types : mutations faux sens, minoritaires (environ une douzaine), délétions, insertions, délétions chromosomiques décalant ou non le cadre de lecture (Abitch et al., 2002 ; Ohno et al., 1995b; Engel et al., 1996b ; Milone et al., 1996; Croxen et al 2002b; Maselli et al., 2002). Les mutations siègent sur l’ensemble du gène codant la sous-unité ε du RACh, les plus nombreuses étant situées dans la région extracellulaire et au niveau de la boucle cytoplasmique entre les domaines transmembranaires M3 et M4 (Brengman et al., 2000; Ohno et al., 1997). Des mutations du promoteur (Nichols et al., 1999; Ohno et al., 1999a) et du peptide signal de la sous-unité ε ont également été décrites (Middleton et al ; 1999). Plus rarement les autres sous-unités du RACh : sous-unité α (Milone et al., 1999), β(Quiram et al., 1999), δ (Ohno et al., 2001) sont impliquées. La prépondérance des mutations de la sous-unité α pourrait s’expliquer par la possibilité de réexpression de l’isoforme fœtale (γ) dans les mutations nulles de la sous-unité ε (Croxen et al., 2001). L’expression clinique de ces SMC n’est pas spécifique et correspond habituellement à une forme caractéristique de SMC. Les anticholinestérasiques et la 3,4-DAP sont efficaces. SMC dus à une anomalie du complexe MuSK-Dok7-rapsyne MuSK est une tyrosine kinase, présente très précocement dans le muscle (dès la prolifération myoblastique) qui active une cascade de signaux impliqués dans tous les aspects de la formation de la jonction neuromusculaire : 1) mise en place de la jonction neuromusculaire; 2) agrégation du RACh par l’intermédiaire de la rapsyne qui assure également l’ancrage du RACh au cytosquelette par via le bêta-dystroglycane ; 3) promotion de la transcription synaptique, en particulier du RACh. MuSK est activée par l’agrine libérée par le motoneurone, mais également par DoK7. Les souris invalidées pour les gènes MuSK ou Dok7 présentent des anomalies majeures de l’innervation et de la différentiation postsynaptique, une absence de clusters de RACh et meurent à la naissance (de Chiara et al., 1996; Okada et al., 2006). Chez les souris invalidées pour le gène de la rapsyne, les myotubes en culture ne forment plus d’agrégats de RACh en présence d’agrine, mais la jonction neuromusculaire est moins perturbée et la transcription des gènes du RACh est conservée (Gautham et al., 1995; Apel et al,1997). Les SMC dus à des mutations du gène de la rapsyne et de Dok7 sont fréquents, plus de 10 % des SMC caractérisés, ceux dus à des mutations de MuSK exceptionnels. SMC dus à des mutations du gène de la rapsyne Les premières mutations du gène de la rapsyne situé en 11p11 ont été identifiées en 2002 (Ohno et al., 2002). Elles concernent un domaine permettant l’auto-association de la rapsyne, étape essentielle pour l’amarrage du RACh au cytosquelette (Ramaro et al., 2001; Ohno et al., 2002a). Ces mutations sont responsables d’une diminution quantitative de la rapsyne et d’une réduction secondaire du RACh à la jonction neuromusculaire. L’hérédité de ce SMC est autosomique récessive. Depuis la première publication portant sur 4 cas, de nombreuses autres observations ont été publiées (Richard et al., 2003; Müller et al., 2003; Dunne et al., 2003; Ohno et al., 2003; Burke et al., 2003; Maselli et al., 2003a; Banwell et al., 2004; Ioos et al., 2004). Plus de 30 mutations ont été rapportées. La mutation N88K est trouvée chez quasiment l’ensemble des patients ; dans la moitié des cas, elle est homozygote. Pour les autres cas, la seconde mutation est localisée tout le long du gène. Les mutations sont de type faux sens dans la moitié des cas. Une microdélétion chromosomique a été rapportée (Müller et al., 2004a). Il existe un effet fondateur pour la mutation N88K (Müller et al., 2004b). Deux phénotypes cliniques sont rapportés : une forme néonatale, voire anténatale avec arthrogrypose, atteinte respiratoire sévère, faiblesse et troubles oculobulbaires majeurs, et des formes légères, plus tardives débutant dans l’enfance, l’adolescence, voire à l’âge adulte. Pour quelques patients aux manifestations initiales très précoces et sévères, l’évolution fut finalement très favorable à l’adolescence (Ioos et al., 2004). La corrélation génotype-phénotype n’est pas simple : dans notre expérience, les mutations homozygotes N88K sont associées à des forme plus légères que les mutations hétérozygotes N88K mais dans la série d’Ohno la sévérité des SMC homozygotes N88K était variable (Ohno et al., 2002). Un tableau très particulier a été rapporté en 1990 chez des patients originaires de la population Juive d’Iraq et d’Iran, qui avaient un phénotype clinique original : SMC bénin avec malformations faciales (prognathisme, face allongée) (Goldhammer et al., 1990). Chez ces patients deux mutations (non N88K) ont été identifiées par la suite dans la région promotrice du gène (Ohno et al. 2003). SMC en rapport avec une mutation du gène Dok7 Okada et coll ont montré en 2006 que les conséquences de l’invalidation du gène Dok7 chez la souris sont majeures avec absence de jonctions neuromusculaires et de clusters de RACh (Okada et al., 2006). Peu après, le groupe de Beeson a rapporté les premiers cas de mutation du gène Dok7 chez 27 patients, issus de 24 familles (Palace et al., 2007) présentant un SMC de transmission récessive, affectant les ceintures. La mutation 1124_1127dupTGCC était présente dans 20 des 24 cas. Deux autres séries ont été publiées : 12 familles et 6 cas isolés (Müller et al., 2007; Anderson et al., 2008). Dans notre groupe, 13 patients ont été identifiés. Les caractéristiques cliniques, analysées sur l’ensemble des cas publiés et notre série, sont les suivantes : début à la naissance dans 1/3 des cas avec hypotonie, difficultés d’alimentation, détresse respiratoire et, dans 2/3 des cas, dans la petite et moyenne enfance, avec faiblesse/fatigabilité des ceintures, difficultés de marche. Pour une petite minorité de patients, l’affection débute à l’adolescence voire chez le jeune adulte. Si dans le SMC Dok7 l’atteinte des ceintures est constante, un déficit distal des extenseurs des doigts est possible, de même qu’un ptosis, une ophtalmoplégie (respectivement 75 et 30 % des cas), une parésie faciale, une atteinte bulbaire avec troubles de déglutition dans 60 % des cas, une atteinte respiratoire chez la majorité des patients, une scoliose évolutive. Les fluctuations sont habituelles, avec des poussées affectant les membres, la déglutition et la respiration, qui peuvent durer plusieurs mois voire plusieurs années. Le décrément est constant à condition de tester des couples nerfs-muscles proximaux. Si le tableau est parfois tardif et bénin, l’évolution est le plus souvent progressive et sévère avec perte de la marche et/ou insuffisance respiratoire requérant une ventilation assistée. Le diagnostic est souvent retardé car la présentation fréquemment très myopathique avec scoliose oriente vers une myopathie congénitale ; une myopathie métabolique est parfois évoquée sur la biopsie du fait de la surcharge lipidique (dans près de la moitié des cas dans notre expérience). Les anticholinestérasiques sont souvent inefficaces voire aggravants. La 3,4-DAP est bénéfique chez les 2/3 des patients, l’éphédrine est également utile. Plus de 25 mutations ont été identifiées tout le long du gène. La mutation 1124_1127dupTGCC quasi constante est soit homozygote soit associée à une autre mutation. Elle est située, comme une dizaine d’autres dans la région C terminale. Neuf mutations sont situées dans le domaine PTB, impliquée dans la phosphorylation de MuSK. Les types de mutation par fréquence décroissante sont les suivants : frameshift > faux sens > non sens et épissage. Dans la série de Muller et coll (Muller et al., 2007), les 2 patients à forme tardive et légère sont homozygotes pour la mutation 1124_1127dupTGCC mais d’autres patients avec cette même mutation, sont plus précocement et sévèrement atteints (Palace et al., 2007). SMC du à des mutations du gène MuSK Un seul cas a été publié par notre groupe (Chevessier et al.,2004). L’observation initiale était celle d’une patiente qui a présenté dans la période néonatale une détresse respiratoire et un ptosis, puis une atteinte très modérée jusqu’à sa première grossesse au cours de laquelle sont survenus des troubles bulbaires et une importante faiblesse des membres. L’association anticholinestérasique-3,4- DAP a été partiellement efficace. Un frère est décédé d’insuffisance respiratoire aiguë à 1 an et demi. L’EMG a révélé un décrément, la biopsie des plaques anormales avec un déficit d’expression de MuSK. Deux mutations, l’une décalant le cadre de lecture dans le domaine extracellulaire IgG like, l’autre faux sens V790M dans le domaine kinase ont été identifiées. Lorsque cette mutation est transférée sur myotubes provenant de souris doublement invalidées pour MuSK, l’agrégation du RACh induite par l’agrine est très réduite. L’expression par électroporation de la mutation sur la patte de la souris reproduit les mêmes anomalies de la plaque motrice que celles de la patiente. Quelques cas de SMC MuSK, de transmission récessive, comme le premier, ont été identifiés mais non encore publiés. Autres SMC postsynaptiques SMC dû à des mutations du canal Sodium SCN4A Il s’agit d’un patient de 20 ans, rapporté par le groupe d’A Engel, présentant depuis la naissance des accès très brefs (3 à 30 mn) de détresse respiratoire et de troubles bulbaires (Tsujino et al., 2003). Le diagnostic a été posé sur l’étude électrophysiologique du muscle intercostal qui révélait une impossibilité d’éliciter un potentiel d’action après stimulation du nerf. Deux mutations du gène SCN4A ont été identifiées, dont l’une située dans le domaine extracellulaire S3/S4, était pathogène lorsque exprimée sur les cellules HEK. SMC par déficit en plectine La plectine est une protéine structurale du cytosquelette hautement conservée. Elle est exprimée dans de nombreux types cellulaires : la peau (au niveau des hémidesmosomes), le muscle (sarcolemme, stries Z) et la membrane postsynaptique. Une observation de déficit en plectine a été décrite chez un patient présentant une myopathie progressive, associée à un syndrome myasthénique (avec atteinte de la face, des membres et de l’oculomotricité, bloc neuromusculaire) et à une épidermolyse bulleuse (Banwell et al., 1999). La 3,4-DAP était efficace, contrairement aux anticholinestérasiques. La physiopathologie de ce SMC reste mal comprise. D’autres cas de plectinopathies ont été rapportés avec atteinte myopathique sans syndrome myasthénique. A la frontière des SMC, l’akinésie fœtale et le pterygium multiple peuvent être dus à des mutations du gène de la sous unité fœtale (γ) du RACh (CHRNG) et de la rapsyne Le syndrome de pterygium multiple (SPM) caractérisé par des palmures du cou, des poignets ou des genoux et des rétractions tendineuses, est soit létal soit non létal, et alors dénommé syndrome d’Escobar. Des mutations de CHRNG ont été identifiées dans 30 % des formes létales de SPM, et également dans certains cas de syndrome d’Escobar (Morgan et al., 2006; Hoffman et al., 2006). La forme fœtale (γ) est remplacée par la forme adulte (ε) à la 32ème semaine, ce qui explique l’akinésie fœtale et les rétractions et, si l’enfant survit, l’absence de symptomatologie myasthénique car la forme adulte du RACh n’est pas affectée. Dans des cas de SPM sans mutation de CHNRG aucune mutation des gènes des sous unité α, β,δ du RACh n’ont été trouvées, mais une mutation homozygote de la rapsyne a été identifiée dans un cas (Vogt et al ; 2008). SMC Synaptique : déficit en AChE Engel et al. ont décrit en 1977 le premier cas de déficit en AChE se caractérisant par l’absence de l’activité enzymatique à la jonction neuromusculaire (Engel et al.,1977). En 1998, simultanément A Engel et col. et Donger et col. ont démontré que le déficit en AChE est lié à des mutations du gène COLQ codant pour la queue collagénique de l’AChE (Ohno et al, 1998b; Donger et al., 1998; Ohno et al., 2000b). A la synapse neuromusculaire, l’AChE est majoritairement sous une forme hétérométrique asymétrique composée d’un à trois homotétramères comportant chacun quatre sous unités catalytiques globulaires, réunies par une queue collagénique (ColQ), elle même de structure trimérique. La forme dominante de l’enzyme est formée de douze sous unités catalytiques. La queue collagénique a pour fonction de concentrer et d’ancrer les sous unités catalytiques à la membrane basale synaptique. Les SMC par mutation du gène ColQ représentent près de 15 % des cas de la série de la Mayo Clinic et dans la série française. Dans une publication récente, 22 nouveaux cas ont été rapportés avec une revue des 38 cas antérieurs (Mihaylova et al., 2008). Les premiers symptômes sont dans 2/3 des cas très précoces, période néonatale ou première enfance : hypotonie, ptôsis, ophtalmoplégie, troubles bulbaires, insuffisance respiratoire avec un risque létal important, un retard des acquisitions motrices. Cependant, des cas de survenue plus tardive, au cours de l’enfance, et peu sévères ont été rapportés, 7 sur 22 dans la série de Mihaylova (Mihaylova et al., 2008). Les symptômes sur l’ensemble des patients publiés sont par ordre décroissant : faiblesse proximale (78,5 %), faiblesse axiale/scoliose (28 %), faiblesse du cou (34,5 %), ophtalmoplégie (30 %), dysphagie/troubles de mastication (24 %), crise respiratoire (35 %) (Mihaylova et al., 2008). Une détérioration progressive survient chez la majorité des patients, parfois tardivement à l’âge adulte. Plusieurs éléments orientent vers ce diagnostic : l’hérédité autosomique récessive, le dédoublement du potentiel d’action lors d’une stimulation unique, l’absence de réponse aux anticholinestérasiques. La lenteur de contraction pupillaire à la lumière est un signe inconstant (environ 1/3 des patients) mais pathognomonique du déficit en AChE. L’étude des plaques motrices permet de poser le diagnostic : AChE qui n’est pas visualisée au niveau des plaques motrices par la technique de Koelle ou le marquage à la fasciculine fluorescente. L’étude morphologique des plaques motrices révèle par ailleurs de franches anomalies dues à l’hyperactivité synaptique induite par l’accumulation d’ACh : altérations focales des replis postsynaptiques avec perte en RACh, dégénérescence sarcoplasmique, petitesse des terminaisons nerveuses (Hutchinson et al., 1993). Plus de 35 mutations récessives ont été décrites, situées tout le long du gène, mais surtout dans le domaine collagénique et la région COOH terminale. Les mutations de la région d’attachement des sous unités catalytiques, appelée PRAD, sont plus rares. La plupart sont originales chez chaque patient, même si quelques unes sont récurrentes : 1082delC, Y430S, T441A (Ohno et al., 1999b ; Müller et al., 2004c; Mihaylova et al., 2008). Elles sont plus souvent homozygotes qu’hétérozygotes et plus souvent de type « tronquant » que faux sens (Ohno et al., 2000b; Mihaylova et al., 2008). Selon leur localisation, les mutations du gène COLQ ont des conséquences différentes : 1) les mutations situées au niveau du domaine N terminal, riche en proline et impliquée dans l’attachement des sous unités catalytiques (région PRAD) empêchent l’accrochage de celles-ci sur ColQ ; 2) les mutations localisées à l’extrémité C terminale de ColQ perturbent l’ancrage de l’enzyme à la membrane basale synaptique ou la trimérisation ; 3) les mutations du domaine collagénique situé entre les 2 extrémités N et C terminales empêchent la trimérisation de ColQ qui ne comprend plus qu’un brin raccourci auquel est fixé un seul tétramère de sous unités catalytiques (Ishigaki et al. 2003 ; Ohno et al., 1999b ; Engel et al. ; 2005). Si généralement on ne peut faire de corrélation entre la sévérité et la localisation de la mutation, voire même pour une même mutation (la même mutation 1082delC homozygote est associée soit à des formes légères, soit sévères), les patients porteurs de la mutation homozygote Y431S ont, à une exception près, une forme légère avec persistance de la forme A12 de l’AChE. Le traitement est difficile car les anticholinestérasiques sont inefficaces voire aggravants ; certains patients ont bénéficié de la 3,4-DAP et de l’éphédrine qui est la molécule la plus efficace (Mihaylova et al. 2008). SMC présynaptiques Ils représentent 7 % des SMC rapportés par la Mayo Clinic. Quatre catégories de SMC présynaptiques ont été rapportés : 1) le déficit en ChAT, le plus fréquent et le seul caractérisé sur le plan moléculaire, 2) le SMC avec pauvreté en vésicules d’ACh, défini sur des critères morphologiques, une seule observation rapportée, 3) le SMC ressemblant à un syndrome de Lambert-Eaton, 4) le SMC avec réduction du nombre de quanta d’ACh., mais sans les caractéristiques électrophysiologiques du syndrome de Lambert-Eaton. Le « SMC avec apnées épisodiques » est lié à des mutations du gène de la ChAT Ohno et coll. ont décrit les premières mutations du gène codant pour la ChAT, molécule présynaptique assurant la catalyse de l’ACh (Ohno et al., 2002b). De transmission récessive, ce SMC débute dans la période néonatale ou dans la petite enfance. Le symptôme le plus caractéristique est la survenue de crises apnéiques (Mora et al., 2007) déclenchées par la fièvre, la fatigue, l’exercice, très brutales et brèves (quelques minutes), volontiers confondues avec des crises comitiales. Les risques sont la mort subite (Byring et al., 2002) ou une anoxie cérébrale par asphyxie trop tardivement prise en charge. Les autres signes sont moins spécifiques : hypotonie, ptôsis, troubles bulbaires. En dehors des poussées, les signes myasthéniques sont souvent modestes voire absents. L’évolution est classiquement favorable avec l’âge, avec une diminution du nombre de poussées mais une proportion significative de patients va développer une faiblesse musculaire croissante, pouvant conduire au fauteuil roulant. Les traitements anticholinestérasiques sont efficaces, en particulier dans la prévention des crises respiratoires. L’EMG révèle un décrément lors de la stimulation répétitive à 3 Hz qui, en dehors des poussées, n’est objectivable qu’après une stimulation à haute fréquence (10 Hz) soutenue pendant 5 minutes (Hart et al., 1979 ; Mora et al., 1987 ). L'étude microélectrophysiologique sur muscle intercostal montre, lors des épreuves de stimulation répétitive à 10 Hz pendant 5 minutes, une diminution d'amplitude des potentiels miniatures et des potentiels de plaque. Ces anomalies sont caractéristiques d'un défaut de resynthèse de l’ACh ou de son empaquetage dans les vésicules synaptiques (Mora et al., 1987). L’histologie du muscle est normale. L’examen ultrastructural montre que les vésicules synaptiques du muscle au repos sont de taille réduite (Mora et al., 1987). Le nombre de RACh, la morphologie postsynaptique, l’activité cholinestérasique sont normales (Mora et al., 1987). Une quinzaine de mutations ont été décrites, situées sur les exons de 6 à 18, épargnant les exons 8,16,17 (Engel et al., 2005). La plupart des mutations sont privées et de type « faux sens » (Ohno et al 2002b; Schmidt et al., 2003 ; Maselli et al, 2003b). La mutation I336T trouvée dans des familles turques pourrait entrer dans le cadre d’un effet fondateur. Comme cela a été montré dans le modèle de souris knock-out (Misgeld et al ., 2002), les mutations entraînent une réduction des capacités catalytiques de l’enzyme allant pour l’une d’entre elles jusqu’à une absence complète d’activité. (Ohno et al., 2002b) Le SMC avec réduction du nombre de vésicules synaptiques Ce type de SMC présynaptique a été décrit chez un seul patient âgé de 23 ans qui présentait un SMC depuis la petite enfance. Dans ce cas, la densité de vésicules synaptiques d’ACh était réduite de 80 % et le nombre de quanta d’ACh libérés très abaissé (Walls et al., 1993). La cause exacte de ce SMC est inconnue. Une anomalie de la synthèse ou du transport axonal de précurseurs des vésicules est envisageable. Autres SMC présynaptiques Deux observations de SMC avec des caractéristiques électromyographiques identiques à celles du syndrome de Lambert-Eaton auto-immun (réduction de l’amplitude des potentiels moteurs, incrément après stimulation à haute fréquence) ont été décrites (Bady et al., 1987; Engel et al, 2003).Dans le cas d’Engel et al,il s’agissait d’un SMC sévère avec hypotonie et insuffisance respiratoire dès la naissance. L’étude microélectrophysiologique révélait une réduction de 90 % du nombre de quanta d’ACh. Le traitement par 3,4-DAP a apporté un bénéfice clinique très modeste malgré l’amélioration des anomalies électrophysiologiques. Aucune mutation n’a été trouvée au niveau des canaux calciques présynaptiques. Quelques autres cas de SMC sporadiques infantiles avec réduction du nombre de quanta d’ACh, mais à la différence du syndrome de Lambert Eaton, sans réduction d’amplitude des potentiels moteurs ni incrément, ont été rapportés en 2001 par le groupe de Maselli, (Maselli et al, 2001) et d’Engel (Milone et al., 2006). Chez un enfant s’ajoutaient des signes d'atteinte du système nerveux central (ataxie cérébelleuse ou nystagmus) (Maselli et al, 2001). Dans aucun de ces SMC présynaptiques ne fut identifiée de mutation tant au niveau du canal calcium CACAPQ que sur d’autres molécules candidates présynaptiques associées à la vésicule ( Maselli et al, 2001; Engel et al, 2003 ; Milone et al, 2006). Syndrome myasthénique des ceintures avec agrégats tubulaires Des agrégats tubulaires ont été rapportés dans des cas de SMC sporadiques ou autosomiques récessifs affectant les muscles des ceintures (Dobkin et al., 1978; Rodolico et al., 2002). Ils diffèrent des SMC dus à des mutations Dok7 par les éléments suivants : absence d’atteinte oculobulbaire, bonne réponse aux anticholinestérasiques, présence d’agrégats tubulaires. Quelques observations de SMC avec agrégats s’associent à une cardiomyopathie (Dobkin et al., 1978; Zephir et al., 2001). Le gène responsable n’est pas connu. Stratégie diagnostique d’un SMC La stratégie comporte 2 étapes complémentaires : 1) diagnostiquer un syndrome myasthénique congénital ; 2) caractériser le type exact du SMC. L’électromyogramme est essentiel révélant un bloc neuromusculaire qui n’est souvent détecté qu’après une étude exhaustive de nombreux couples nerfs-muscles, en particulier proximaux qui peuvent être les seuls à présenter un trouble de neurotransmission. Une stimulation prolongée de 5 minutes à 10 Hz avant la stimulation classique à 3Hz peut être nécessaire pour faire apparaître le décrément, en particulier dans le déficit en ChAT. Un second argument électromyographique est capital : la réponse répétitive après stimulation unique chez un patient non traité par les anticholinestérasiques ; cet aspect, qui traduit un hyperfonctionnement pathologique de la transmission neuromusculaire, signe un SMC et oriente vers 2 étiologies de SMC : le syndrome du canal lent ou un déficit en AChE. Souvent le diagnostic est délicat : survenue tardive (adolescence voire à l’âge adulte), absence de réponse aux anticholinestérasiques, absence d’histoire familiale, expression myopathique au premier plan avec faiblesse permanente non ou peu fluctuante, atrophie, scoliose, rétractions, EMG myogène, sans bloc neuromusculaire sur les troncs distaux, biopsie trompeuse car révélant des anomalies franches (le plus souvent : prédominance1, atrophie 2, surcharge lipidique). Les diagnostics erronés les plus fréquents sont : une myopathie congénitale, une myopathie métabolique (en cas de surcharge lipidique), une myasthénie auto-immune si l’affection apparaît au-delà de la petite enfance. Il existe d’exceptionnels cas de myasthénie auto-immune à début fœtal avec arthrogrypose et déficit très sévère lorsque la mère transfère des anticorps dirigés contre le RACh fœtal. Les symptômes myasthéniques manquent souvent chez la mère mais la mise en évidence d’anticorps anti-RACh chez la mère et l’enfant à la naissance confirmera le diagnostic. Pour ce qui concerne le diagnostic étiologique, certaines entités sont facilement suspectées : 1) le syndrome du canal lent (transmission dominante, dédoublement du potentiel moteur, inefficacité des anticholinestérasiques), 2) le déficit en AChE (transmission récessive, dédoublement du potentiel moteur, inefficacité des anticholinestérasiques, contraction pupillaire très lente à la lumière), 3) les SMC dus à une mutation fondatrice de la sous unité ε du RACh si celui-ci survient dans la population gitane ou d’Afrique du nord (voir infra). Deux autres SMC peuvent être évoqués sur des caractéristiques particulières : le déficit en ChAT suspecté devant des épisodes soudains et brefs dominés par des crises apnéiques contrastant avec un examen intercritique quasi normal et un décrément n’apparaissant qu’après effort ou stimulation à haute fréquence et un déficit primaire en rapsyne sur présence de signes fœtaux et de rétractions, en particulier des doigts. En l’absence des éléments d’orientation décrits plus haut, l’étude génétique moléculaire est entreprise, en débutant par les 3 gènes les plus souvent impliqués : sous unité ε du RACh, rapsyne et DoK7. Si cette première série de gènes est éliminée, les 7 autres gènes seront étudiés systématiquement. La place de la biopsie musculaire s’est réduite. La biopsie deltoïdienne avec étude des plaques motrices étudiées permet de diagnostiquer un déficit en AChE (déficit majeur de l’enzyme avec la technique histochimique de Koelle ou avec la fasciculine fluorescente). Une perte en RACh et une expression de la sous unité fœtale γ du RACh s’inscrivent en faveur d’une mutation de la sous unité ε. Peu d’équipes possèdent la technicité pour l’étude micoélectrophysiologique du muscle intercostal ou de l’anconé qui est particulièrement utile en cas de suspicion d’un SMC présynaptique avec réduction du nombre de quanta d’ACh. L’étude des plaques motrices est surtout précieuse en cas de SMC non identifié : la constatation d’un déficit par immunomarquage d’une des molécules de la jonction neuromusculaire aura une valeur d’orientation même si celui-ci peut être secondaire. Si le SMC n’est toujours pas caractérisé, on proposera une étude de liaison, si la famille est informative, ou de gène candidats éventuellement suspectés sur la biopsie. Quand une nouvelle mutation d’un gène connu ou un nouveau gène est identifié, il faudra valider le caractère pathogène de la mutation soit dans un système d’expression cellulaire (cellules HEK, oocyte…) soit chez l’animal (souris exprimant la mutation). Corrélations génotype-phénotype et pronostic Les corrélations phénotype-génotype sont complexes. Des manifestations cliniques identiques ont été retrouvées dans des SMC dus à des gènes différents : si les apnées épisodiques sont évocatrices de mutations du gène de la ChAT, elle sont également décrites dans des SMC dus à des mutations du gène de la rapsyne, de l’acétylcholinestérase et de la sous unité δ du RACh. L’arthrogrypose fréquente dans les mutations du gène de la rapsyne est également présente dans les mutations de la sous unité δ du RACh (Bronlow et al., 2001). L’atteinte prédominante des ceintures est trouvée dans les SMC dus à des mutations des gènes DoK7, Colq et dans les SMC avec agrégats tubulaires. La même double mutation N88K de la rapsyne est associée à des SMC sévères ou bénins. Une variabilité intrafamiliale n’est pas rare dans les SMC. Le pronostic des SMC n’est pas facile à poser : amélioration dans des formes initialement sévères de SMC (en particulier en cas de mutations du gène de la rapsyne), aggravation tardive avec recours au fauteuil roulant et à la ventilation assistée dans des SMC dus à des mutations de gènes variés : DOK7, RAPSN, COLQ. Le schéma évolutif peut se modifier au cours de l’évolution : évolution par poussée faisant place à une évolution progressive. Traitement Des mesures non spécifiques sont indispensables : prise en charge d’une insuffisance respiratoire par ventilation assistée, des troubles de déglutition, d’une scoliose sévère et des rétractions, respect des contre-indications médicamenteuses (voir tableau). La corticothérapie, les immunosuppresseurs, les immunoglobulines intraveineuses ou les plasmaphérèses n’ont aucune place dans le traitement des SMC même si, pour des raisons inconnues, certains patients, initialement considérés comme des myasthénies auto-immunes séronégatives, ont partiellement et transitoirement répondu à ces traitements. Les anticholinestérasiques sont efficaces dans la plupart des SMC, y compris dans le déficit en ChAT, à l’exception de trois catégories de SMC : déficit en AChE, syndrome du canal lent et SMC par mutation de DOK7. La 3,4-DAP, dont le mode d’action est présynaptique (libération accrue de vésicules d’ACh), n’est pas seulement efficace dans les SMC présynaptiques avec réduction de la libération d’ACh mais elle est souvent bénéfique dans les SMC postsynaptiques (déficit en AChR sans anomalies kinétiques, SMC par mutation de la rapsyne et Musk) (Harper et al, 1998). L’effet de la 3,4-DAP et des anticholinestérasiques se potentialise souvent. Les patients présentant un syndrome du canal lent répondent favorablement aux molécules réduisant le temps d’ouverture du RACh : quinidine, 200 mg, 3 fois par jour chez l’adulte, et à la fluoxetine à la dose de 80 à 120 mg par jour (Harper et al., 2003). L’éphedrine, dont le mécanisme d’action n’est pas clair, a un effet positif dans les SMC dus aux mutations de DOK7 (Palace et al.2007, Mihaylova et al., 2008) et dans le déficit en AChE (Mihaylova et al., 2008). Pour le déficit en AChE, la 3,4-DAP est parfois efficace. Une revue récente a été consacrée au traitement des SMC (Engel et al., 2007) Un diagnostic prénatal est possible lorsque le gène a été caractérisé. Conclusions Dans les deux dernières décennies, des progrès remarquables ont été obtenus dans la connaissance des SMC, portant sur leur caractérisation phénotypique, leur diagnostic moléculaire et leur traitement. L’épidémiologie des SMC est mieux connue et il apparaît que leur fréquence a été sous estimée du fait de nombreux cas non correctement diagnostiqués. Un diagnostic prénatal peut être maintenant proposé. Les travaux futurs devront permettre d’identifier les gènes impliqués dans les SMC non encore étiquetés, représentant encore la moitié des cas. Ils nécessiteront une collaboration internationale entre cliniciens, morphologistes, généticiens et neurobiologistes.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :