Publié le 09 jan 2012Lecture 12 min
Syndrome du QT long congénital : quand le rechercher ?
V. LUCET - Unité de rythmologie pédiatrique, Château des Côtes, Les Loges-en-Josas

Le syndrome du QT long congénital (SQTL) est une affection fréquente. Sa prévalence génétique est estimée à 1/2 000 individus. C’est aussi une affection grave puisqu’il expose à la survenue de troubles du rythme paroxystique à type de torsades de pointe, pouvant conduire à la mort subite.
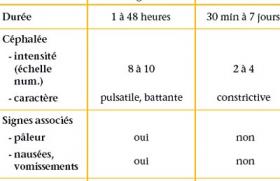
Le diagnostic de cette affection familiale est souvent difficile puisque la seule mesure de QT corrigé expose à un nombre élevé de faux positifs ou, à l’inverse, de faux négatifs. La biologie moléculaire est donc devenue l’une des méthodes de référence pour confirmer le diagnostic, mais il s’agit d’explorations complexes, longues et qui comportent aussi leurs parts d’incertitude. En dehors des cas où un QT manifestement trop long est découvert de façon fortuite, à l’occasion d’un ECG systématique, la recherche d’un syndrome de Romano-Ward ou de Jervell (avec surdité) s’inscrit dans un contexte : le plus souvent, il s’agit de l’exploration d’un malaise ou d’un équivalent convulsif. La recherche d’un QT long congénital doit aussi être effectuée dans le cadre d’une enquête familiale, après découverte d’un cas index. Parfois, c’est en présence d’une bradycardie anormale qu’il faut savoir rechercher un QT long, enfin, après le drame que constitue la mort subite inexpliquée d’un nourrisson ou d’un grand enfant, la recherche d’une arythmie génétique s’impose. QT long et canalopathies rythmiques Le syndrome de QT long est cliniquement et génétiquement hétérogène. Les progrès de la biologie moléculaire ont permis la découverte d’un nombre croissant de mutations. À ce jour, douze gènes ont été identifiés, mais trois gènes principaux rendent compte de la majorité des cas : le gène KCNQ1 pour le SQTL de type 1 (LQT1), le gène KCNH2 (HERG) pour le SQTL de type 2 (LQT2) et le gène SCN5A pour le SQTL de type 3. KCNQ1 et KCNH2 codent pour une sous-unité des canaux potassiques, alors que SCN5A code pour la sous-unité alpha des canaux sodiques cardiaques. Bien d’autres mutations ont été identifiées, notamment sur des gènes régulateurs, si bien que tout peut se voir en matière de mutation et notamment, des statuts d’homozygotes ou d’hétérozygotes composites, ainsi que des mutations doubles réalisant alors souvent des formes graves de QT long. Quoi qu’il en soit, ce syndrome hétérogène du QT long (qu’il s’agisse du syndrome dominant de Romano-Ward, ou récessif de Jervell avec surdité) s’inscrivent maintenant dans le cadre plus large des canalopathies rythmiques (1), au même titre que les tachycardies ventriculaires polymorphes qui associent un QT normal et des arythmies ventriculaires polymorphes d’effort ou de stress pouvant conduire à la mort subite et liées à des mutations génétiques régulant les courants calciques. De la même façon, le syndrome du QT court (QTc < 300 ms) avec risque élevé de mort subite est lié à des mutations spécifiques dans HERG, KCNQ1 ou KCNJ2. Enfin, et pour ne citer que les plus fréquentes des canalopathies rythmiques, les syndromes de Brugada, rares chez l’enfant, associent un sus-décalage en « dôme » du segment ST en précordiale droite, et un risque de mort subite par fibrillation ventriculaire lié à une mutation dans le gène SCN5A. Diagnostic clinique Faire le diagnostic de SQTL, c’est évidemment commencer par la mesure de l’intervalle QT dans les dérivations des références (D2 ou V5). Une durée du QT corrigé par la fréquence cardiaque QTc (selon la formule de Bazett : QTc [en secondes] divisé par la racine carrée du RR précédent [en secondes]) > 440 ms est associée à une forte probabilité diagnostique ; cependant, le QTc varie d’un moment à l’autre chez le même individu, et l’on ne peut formellement éliminer le diagnostic en cas d’ECG normal dans un contexte évocateur. Il a été montré qu’il existe un chevauchement important des intervalles QTc chez les sujets sains et chez les sujets génétiquement atteints : on estime en effet que 25 % environ des QT longs génétiques ont un QTc < 440 ms, alors que 25 % des nourrissons sains ont un QTc > 440 ms. C’est pourquoi, nous utilisons les critères établis par l’équipe de Lariboisière : le diagnostic de QT long paraît probable si le QTc est > 460 ms ou si le QTc est > 440 ms et qu’il existe une anomalie morphologique de l’onde T ou une bradycardie insolite. Le diagnostic doit aussi être suspecté, même si le QT est normal, devant une syncope ou une torsade de pointe dans une famille de QT long. Quoi qu’il en soit, en cas de doute, il faut savoir effectuer un ECG à toute la famille, couplé à une enquête moléculaire (après consentement éclairé des patients). On ne peut formellement éliminer le diagnostic en cas d’ECG normal dans un contexte évocateur. QT long et malaises La possibilité d’une arythmie de type torsade de pointe en rapport avec un syndrome de QT long doit être évoquée chez l’enfant devant tout malaise et conduire à la réalisation d’un ECG, couplé, en cas de syncope authentique, avec un enregistrement de Holter et/ou une épreuve d’effort(2). En effet, la mutation la plus fréquente retrouvée (LQT1) donne essentiellement des syncopes déclenchées par l’effort ou l’émotion. Dans ce cadre, la survenue d’un accident de baignade ou d’une noyade récupérée est très évocatrice : l’équipe de la Mayo Clinic retrouve 11 % d’accidents de baignade sur une série de 388 cas index génétiquement prouvés, dont 85 % de LQT1. Toute noyade récupérée doit donc donner lieu à une exploration rythmologique complète à la recherche d’un QT long (ou d’une TV polymorphe). Les SQTL de type 2 donnent volontiers des syncopes déclenchées par le stress, et en particulier les stimulis auditifs. Enfin, les LQT3 donnent plus volontiers des syncopes au cours du repas ou du sommeil. QT long et enquête familiale La découverte d’un cas index doit conduire à une enquête familiale complète avec ECG et prélèvement génétique. Il paraît aussi utile de réaliser un ECG à toute la famille d’un cas suspect, affichant un QT limite et/ou une histoire de syncope. La découverte, dans la fratrie, ou chez les parents, d’un deuxième cas suspect ou d’un QT franchement augmenté permet alors d’étayer le diagnostic du cas index et de proposer l’enquête moléculaire. La réalisation systématique de ces enquêtes rend compte de la découverte d’un nombre élevé de QT long asymptomatique ou méconnu : on estime actuellement que 60 % des enfants porteurs d’un QT long sont découverts à l’issue de l’enquête familiale menée à partir de 40 % des cas index. Cette population de QT long issue des enquêtes familiales est constituée d’enfants plus jeunes, ayant des QT statistiquement moins longs et un pronostic en règle plus favorable que les index (3). Figure. Syndrome du QT long : démarrage d’une torsade de pointe à l’occasion d’une extrasystole. La découverte d’un cas index doit conduire à une enquête familiale complète avec ECG et prélèvement génétique. QT long et épilepsie Les troubles du rythme ventriculaires paroxystiques du syndrome du QT long peuvent s’exprimer (comme les TV catécholergiques) sous la forme d’un malaise convulsif. Cela conduit bien souvent à porter à tort le diagnostic d’épilepsie et contribue à un retard dommageable à la mise en route d’un traitement spécifique par bêtabloquant. Dans une série de 31 cas de QT long recensé en Nouvelle-Zélande entre 2000 et 2005, les auteurs(4) constatent un retard moyen au diagnostic de 2,5 ans après la première syncope dans 13 observations. Dix de ces patients ont « bénéficié » d’un EEG qui a conduit à porter à tort le diagnostic d’épilepsie dans un cas sur deux. Pour ces 5 cas, le retard diagnostique a été prolongé (9,5 ans) rendant compte de la survenue de 4 morts subites dans ces familles. Il apparaît donc essentiel de pratiquer un ECG avec mesure du QTc devant la survenue d’une première crise d’épilepsie. Les anomalies de l’EEG doivent être confirmées à distance de la crise initiale, et le diagnostic doit être reconsidéré en cas de résistance au traitement anticomitial. Cette méconnaissance du diagnostic du QT long n’explique cependant vraisemblablement pas toutes les morts subites constatées dans l’épilepsie essentielle (jusqu’à 18 % des cas selon les séries). Récemment, il a été montré chez la souris(5), une co-expression à la fois dans le cerveau et dans le coeur d’une mutation KCNQ1. Il apparaît donc essentiel de pratiquer un ECG avec mesure du QTc devant la survenue d’une première crise d’épilepsie. QT long et bradycardie Une bradycardie sinusale insolite est retrouvée dans environ 25 % des cas, particulièrement chez les sujets jeunes ayant une mutation dans LQT3. Le bloc auriculo-ventriculaire (le plus souvent 2/1) compliquant un syndrome de QT long a un pronostic péjoratif. Il est le plus souvent constaté chez le foetus ou au cours du premier mois de vie. Ces formes très graves de QT long néonatals sont en rapport le plus souvent avec une mutation dans HERG ou avec des formes homozygotes ou hétérozygotes composites. QT long et surdité Le syndrome de Jervell associe une surdité bilatérale, congénitale, profonde d’origine neurosensoriel et un QT parfois très prolongé. Cette affection autosomique récessive est liée à des mutations dans KCNQ1 et KCNE1. De fait, les deux parents sont porteurs d’une mutation à l’état hétérozygote et le plus souvent asymptomatique ; leurs enfants homozygotes, en revanche, présentent des formes graves de QT long avec un QTc souvent > 500 ms et la survenue de syncopes précoces (dans un cas sur deux avant 3 ans). En l’absence de traitement, 50 % des sujets atteints vont décéder avant l’âge de 15 ans(6). C’est dire l’importance du dépistage de la surdité chez les nouveau-nés. Cette surdité profonde est liée à un défaut de production d’endolymphe dans la cochlée, et est susceptible d’être traitée par des implants cochléaires. QT long et mort subite C’est à P. Schwartz (7) que revient le mérite d’avoir évoqué un lien entre QT long et mort subite du nourrisson (MSN). Dès 1982, en effet, il publiait une série de 2 000 nouveau-nés documentés par un ECG à la naissance et suivi jusqu’à l’âge de 1 an. Trois d’entre eux devaient décéder de mort subite, leur QTc mesuré à la naissance était prolongé (> 2 DS). Seize ans plus tard, le même auteur reprenait, en l’augmentant, la même étude à propos, cette fois-ci, de 33 000 nouveau-nés. Un an plus tard, 24 d’entre eux avaient faits une mort subite et 12 présentaient un QTc > 440 ms. À la suite de cette étude, P. Schwartz a proposé en Italie, le dépistage systématique du QT long par la réalisation d’un ECG à la naissance. Ce protocole est cependant discuté, car il se heurte à de nombreuses difficultés d’interprétation du QT à la naissance, avec souvent des ondes T peu amples et très variables d’un jour à l’autre. Cela majore considérablement le nombre de faux positifs ou de faux négatifs déjà élevé dans la population générale. Enfin, les conséquences psychologiques, médicolégales et économiques d’une telle recherche ont fait renoncer la plupart des pays à appliquer ce protocole. Pourtant, le même auteur rapportait en 2000 le premier cas prouvé de MSN liée à une mutation en SCN5A. D’autres publications ont suivi confirmant le risque de mort subite au cours du sommeil chez les sujets porteurs d’un LQT3. Depuis, de nombreuses études de biologie moléculaire réalisées en post mortem chez des nourrissons ayant fait une MSN ont permis de découvrir des mutations dans les 5 principaux gènes de SQTL. C’est le mérite, notamment, des travaux de la Mayo Clinic (8), d’avoir étendu ces recherches aux morts subites du grand enfant et aux autres canalopathies rythmiques ; si bien qu’actuellement on estime que ce type de mutation pourrait être responsable d’environ 10 % des MSN, et même du tiers des morts subites du grand enfant à autopsie négative. Ces données incitent donc maintenant à pratiquer un examen cardiologique complet aux apparentés du 1er et du 2e degré d’un enfant décédé de mort subite avec une autopsie normale. De la même façon, une autopsie moléculaire paraît justifiée devant toute MSN quand l’enquête étiologique, les prélèvements post mortem et l’autopsie complète n’ont pas permis de conclure (9). Points forts ● Diagnostic probable d’un QT long : – QTc > 460 ms ; – QTc ≥ 440ms, avec anomalie de la forme de l’onde T ou bradycardie ; – syncope ou torsade de pointe dans une famille de QT long. ● Phénotype clinique : – LQT1 : syncope d’effort ou d’émotion ; – LQT2 : syncope au stress (auditif) ; – LQT3 : syncope au repos ou au sommeil. ● QT long et mort subite : Intérêt de faire : – un ECG à toute la famille (± Holter) ; – une autopsie moléculaire (si autopsie normale). Il paraît licite de pratiquer un examen cardiologique complet aux apparentés du 1er et du 2e degré d’un enfant décédé de mort subite avec une autopsie normale. En pratique, on retiendra Il faut savoir penser au syndrome de QT long devant tout malaise ou syncope de l’enfant, devant un accident de la baignade ou une épilepsie résistante au traitement. L’attention peut aussi être attirée par une bradycardie insolite, notamment chez le nouveau-né ou plus rarement par une surdité congénitale profonde. Compte tenu du pronostic vital, tout retard diagnostique ou toute méconnaissance peut être catastrophique. Pourtant, affirmer un QT long est souvent difficile, surtout dans les formes frontières où le QTc avoisine les 440 ms. En cas de doute, il faut étendre l’enquête à toute la famille, répéter les ECG et compléter le bilan par un enregistrement de Holter et/ou une épreuve d’effort. L’apport de la biologie moléculaire peut être décisif, mais a ses propres limites.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :